Opinion
Prediction of protein Post-Translational Modification sites: An overview

Md. Mehedi Hasan1* and Mst. Shamima Khatun2
1Department of Bioscience and Bioinformatics, Kyushu Institute of Technology, 680-4 Kawazu, Iizuka, Fukuoka 820-8502, Japan
2Laboratory of Bioinformatics, Department of Statistics, University of Rajshahi, Rajshahi, Bangladesh
*Address for Correspondence: Md. Mehedi Hasan, Department of Bioscience and Bioinformatics, Kyushu Institute of Technology, 680-4 Kawazu, Iizuka, Fukuoka 820-8502, Japan, Email: [email protected]
Dates: Submitted: 27 February 2018; Approved: 01 March 2018; Published: 02 March 2018
How to cite this article: Hasan MM, Khatun MS. Prediction of protein Post-Translational Modification sites: An overview. Ann Proteom Bioinform. 2018; 2: 049-057. DOI: 10.29328/journal.apb.1001005
Copyright License: © 2018 Hasan MM, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Introduction
Post-translational modification (PTM) refers to the covalent and enzymatic modification of proteins during or after protein biosynthesis. In the protein biosynthesis process, the ribosomal mRNA is translated into polypeptide chains, which may further undergo PTM to form the product of mature protein [1]. PTM is a common biological mechanism of both eukaryotic and prokaryotic organisms, which regulates the protein functions, the proteolytic cleavage of regulatory subunits or the degradation of entire proteins and affects all aspects of cellular life. The PTM of a protein can also determine the cell signaling state, turnover, localization, and interactions with other proteins [2]. Therefore, the analysis of proteins and their PTMs are particularly important for the study of heart disease, cancer, neurodegenerative diseases and diabetes [3,4]. Although the characterization of PTMs gets invaluable insight into the cellular functions in etiological processes, there are still challenges. Technically, the major challenges in studying PTMs are the development of specific detection and purification methods.
The PTMs of proteins have been detected by a variety of experimental techniques including the mass spectrometry (MS) [5,6], liquid chromatography [7], radioactive chemical method [8], chromatin immune precipitation (ChIP) [9], western blotting [10], and eastern blotting [7]. The MS technique is one of the mainstay routes in detecting PTMs in a high-throughput manner. The new MS and capillary liquid chromatography instrumentation have made revolutionary advance in enrichment strategies in our growing knowledge of various PTMs [11]. The last decade of the actual description of many PTMs complexity has emerged through the diverse technologies and thousands of precise modification sites can now be identified with high confidence [12-20]. A similar strategy of fragmentation for PTM identification is the beam-type collision-induced dissociation, also called higher energy collisional dissociation [21]. These types of fragmentation are characterized by the higher activation energy. Most of the fragmentation methods of precursor ions are based on the radical anions or thermal electrons [22]. These methods are advantageous over collisionally activated dissociation methods for detecting the unstable PTMs (e.g. O-GlcNAc and phosphorylation), due to the peptide support fragmentation method is effectively independent of the amino acid sequence [23-25]. To date, more than 350 types of PTMs have been experimentally discovered in vivo [26]. The common PTMs are phosphorylation, ubiquitination, succinylation, acetylation, pupylation, sumoylation, glycosylation, and so on. In addition, pupylation referring to the modification of lysine residues with a prokaryotic, ubiquitin-like protein (i.e. Pup) is another PTM in bacteria.
In general, the experimental analysis of PTMs often requires labor-intensive sample preparations and hazardous or expensive chemical reagents. For instance, in the radioactive assay in the kinase based methods are often separated from non-radioactive ATP by the kinase assay and generates radioactive waste [8]. Since most of the radioactive substance deal a short half-life, the fresh reagent must be frequently required for identifying PTMs. And sometimes, the substrate concentration of assay is often much higher than the expected substrate concentrations [27]. In summary, the identification of PTMs by the experimental techniques is laborious, time-consuming and usually expensive. As an alternative, the computational methods are more efficient for identifying large-scale novel PTM substrates.
The last several decades have been remarkable progress in the identification and functional analysis of PTMs in proteins. The PTMs play a vital role in protein folding, protein function, and interactions with other proteins [28,29]. Due to the important biological functions of protein PTMs, it is very important to analyze and understand the function of PTMs. In contrast to the traditional experimental methods, computational analysis of PTMs has also been an attractive and alternative approach due to its accuracy, cost-effective and high-speed. The computational tools can narrow down the number of potential candidates and rapidly generate useful information for investigating further experimental approach. Thus far, the prediction of protein PTMs is an important research topic in the field of protein bioinformatics. Although the great progress has been made by employing various feature representation and statistical learning approaches with numerous feature vectors, the problem is still far from being solved. An overview of protein PTM sites prediction is presented in figure 1.
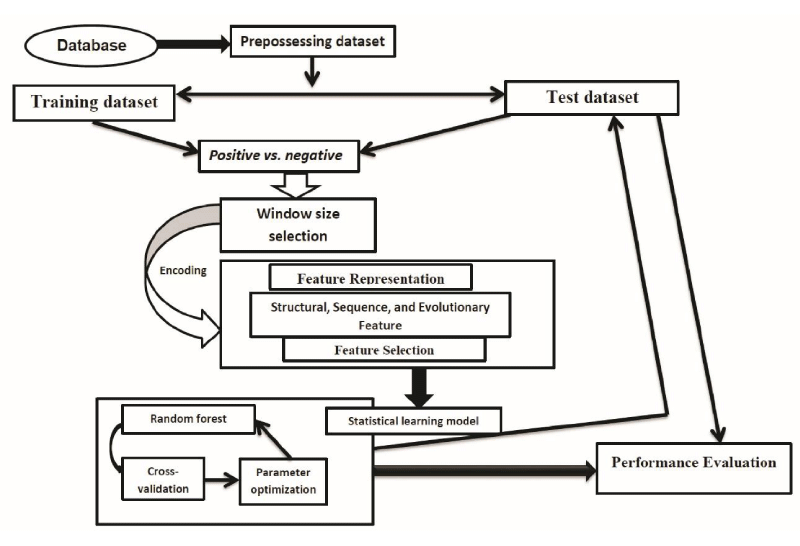
Figure 1: A brief flowchart template for computational prediction PTM sites. Firstly, the dataset was collected from the published database. Secondly, then need to be preprocessed the collected datasets for making proper positive and negative samples. Thirdly, the resulting encoded feature vectors were independently put into the statistical learning models to produce independent prediction scores. Finally, optimum performance scores were calculated by using cross-validation and parameter optimization, a confident cutoff was considered to identify the PTM site.
Feature representation
Feature representation is one of the most important steps for predicting PTM sites. Suitable features in the prediction model allow the precise prediction of protein PTMs. In general, these features refer to the description of the sequences and local structures around these protein functional sites. Ideally, the features can clearly distinguish PTM sites from the random modification residues. In the real world, however, the feature of protein functional sites can also exist on the non-functional sites of proteins. In the prediction PTM sites, this specific problem is particularly prominent due to the sequence diversity. For instance, some motifs are very weak and some are not available without the sequence evolutionary information [30-35]. To address this problem, we can search PSI-BLAST [36-38] against the NCBI NR database to generate a profile (i.e. position-specific scoring matrix (PSSM)). Such sequence profiles reflect the conservation and variation between protein sequences through the evolutionary information [39-42].
There are also numerous protein structure features proposed. For example, one can examine the amino acid solvent accessibility of PTM sites. Examining the residue interactions that uphold the stability of protein structures (including electrostatic interactions, hydrophobic interactions, van der Waals interactions, disulfide bonds, hydrogen bonds, and so on) may be also helpful [43,44]. Additionally, the residues’ structural flexibility information like root mean square deviation and B -factor is sometimes useful, too. Last but not least, some of the residue contact network parameters (betweenness, closeness, degree, and clustering coefficient) were used as features for protein PTM prediction [45]. In a real-world prediction task, note that the scientists usually use the integrated feature set to identify the protein PTM sites.
The statistical algorithm of PTM sites prediction
After determining the appropriate features, the next job is to use an appropriate machine learning algorithm to integrate these features for the prediction of protein PTM sites. It will improve the accuracy of the prediction if the prediction algorithm is appropriate. These prediction algorithms of PTM sites can be classified into two categories, i.e. statistical probabilistic algorithms and machine learning algorithms. In following, we will discuss some of these algorithms.
Naïve bayes
Naïve Bayes is a predictive algorithm based on the statistical learning theory of Bayesian theorem. The advantage is that this algorithm is easy and simple to calculate. In Bayesian theorem, the posterior probability of a random event is the conditional probability, which is assigned after the relevant evidence been taken into account. Bayesian assumes that a property of a given value is affected by the other values. This assumption is not often established on the model, so its accuracy can be rejected for other properties of the class forecasting models, such as linear regression and logistic regression models. The majority of biologists think that for analyzing the biological data Naïve Bayes is an important algorithm [46]. Although, these methods affected by many outlier and do not handle the noise model [47]. In bioinformatics research, Naïve Bayes algorithms are widely used [48-50].
From more than 20 years ago, machine learning algorithms have been widely used for an interdisciplinary field. There are related to the probability theory, approximation theory, convex analysis, complexity theory and other disciplines. To predict the unknown data, they have been widely used (http://en.wikipedia.org/wiki/Machine_learning). Since machine learning algorithms are highly automated, accurate and predictive, they have a very wide range of applications, such as the data mining, computer vision, natural language processing and biometrics. Although the performance of machine learning models shows a very good accuracy, they do not help the researchers to understand the deep mechanisms and biological significance [51]. Thus, sometimes the machine learning methods are criticized as the “black box” learning.
In early 1959, Arthur Lee Samuel defined the machine learning as “the field of study that gives computers the ability to learn without being explicitly programmed” [52]. For the prediction of protein PTMs, some common machine learning algorithms are widely used such as support vector machine (SVM), artificial neural network (ANN) and random forest (RF). Subsequently, we will discuss these three common machine learning algorithms.
Random forest
RF is an ensemble supervised learning algorithm [53]. It can integrate multiple classifiers to improve the performances of the prediction [54-56]. It is well known that for a supervised classifier, the model classification error is partly attributed to the different distributions between the training and the unknown samples (Figure 2A). In contrast, if sets have contained a certain degree of disturbance to the training set, which can determine the more general prediction and it can also remove the bias of a single classifier [57-58]. Several advantages of RF are as follows: 1) For the reliable individuals characteristic, RF can produce a highly accurate classifier. 2) It can handle a large number of input variables. 3) It can produce the importance of variable from a given class variable. 4) In the construction of the forest, it does not produce any bias results. 5) It contains a good way to estimate the loss or missing of data and if a large part of the information is lost, it can still maintain accuracy. 6) For the unbalanced classification problem, it can take balance errors. 7) It can calculate the degree of intimacy in each case, such as in data mining for detecting the deviations (outlier) and it is also very useful for data visualization. 8) It can also be used in the extended unlabeled dataset, such as non-supervised or supervised clustering. 9) The learning process is very fast than other algorithms. It has a high predictive accuracy, good tolerance of outliers and noise. It has been widely used in the field of bioinformatics research [59-63].
Support vector machine
In 1995, SVM was firstly proposed by Corinna and Vapnik [64], which can solve the nonlinear and multidimensional pattern recognition problem. It uses a nonlinear transformation method and transforms low-dimensional data to high-dimensional feature space. It can look for a hyperplane in a high-dimensional space to maximize the margin between two types of data (Figure 2B). In other words, as long as suitable kernel functions, SVM can solve the high-dimensional classification problem. In the theory of SVM, SVM with different kernel functions has led to different algorithms. The most commonly used SVM is radial basis function (RBF) kernel. Until now, many types of SVM software packages have been developed, such as SVM-Light (http://svmlight.joachims.org/), LIBSVM [65], Gist [66], Weka [67], and so on.
Recently, in bioinformatics research, SVM has been widely used in various topics, including protein PTM prediction [38,68,69], protein residue contact prediction [70], protein fold recognition [71], protein secondary structure prediction [72], etc.
Artificial neural network
In 1969, after the publication of machine learning research by Marvin and Seymour the neural network research has been boomed [73]; they initially discovered the two key issues with the computational machines learning neural networks. The first one was the single-layer neural networks for processing on the circuit area. The second was the significant issue of computers for processing the power to effectively handle the long run time by large neural networks.
In machine learning and cognitive science approaches, the ANN is a family of statistical learning models and it is inspired by the biological neural networks (central nervous system of animals, in particular, the brain). This learning algorithm is used to estimate the approximate functions of input samples. ANN is also presented as systems of interconnected “neurons” which can exchange the messages between each other. The connections are generally numeric weighted and it can be tuned based on the internal experience. In general, ANN consists of three layers: input layer, hidden and output layer (Figure 2C). The potential law is needed for analyzing the independent variables and dependent variable in the ANN, which can calculate the new input data [74].
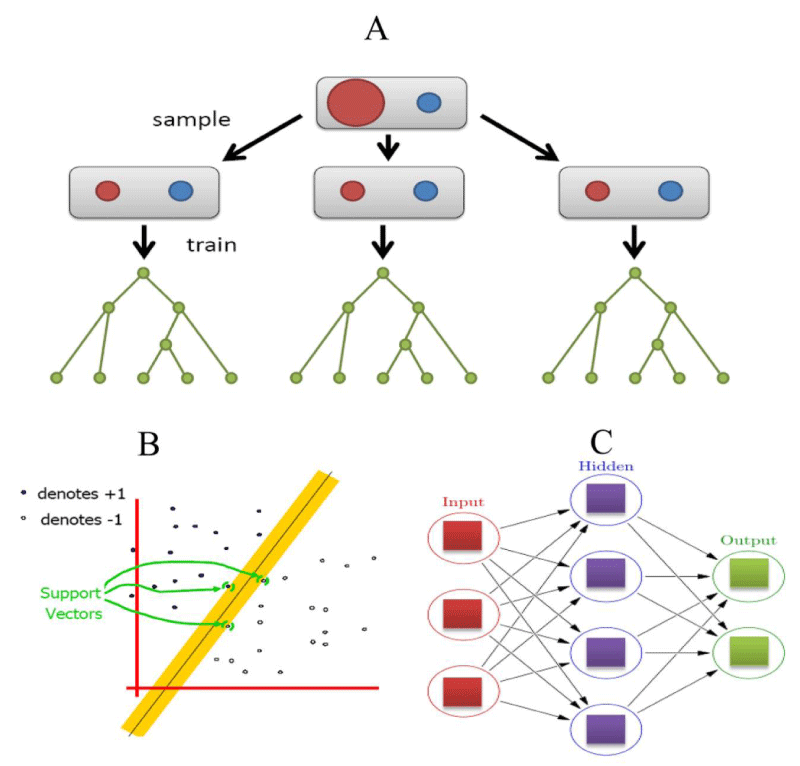
Figure 2: Machine learning algorithms for PTMs prediction. (A) Random forest classifier. Here, red color denotes training samples, while green color denotes testing samples and each node represents a tree. (B) SVM classifier and (C) artificial neural network. Here, each circular node represents an artificial network, while an arrow represents a connection from the output of one network to the input of another.
In the field of bioinformatics, ANNs have also a wide range of applications, such as protein functional sites prediction [75-77], protein secondary structure prediction [78,79] and tertiary structure prediction [80]. Common implementations of ANN software are FANN (http://leenissen.dk/Fann/WP/) and SNNS (http://www.ra.cs.uni-tuebingen.de/SNNS/).
In summary, machine learning algorithm is a subfield of computer science and statistics that evolved the study of pattern recognition and computational learning theory in artificial intelligence. For PTM prediction machine learning algorithm is an essential step for testing the model performance.
Conclusions
The expansion and application of PTM site prediction are emerging as a promising field in protein bioinformatics research. High-throughput omics-based techniques have been widely used in the study of PTMs. For our better understanding the function of PTMs, more accurate computational analysis is required. Combining experimental and computational schemes will certainly accelerate our knowledge by analysis of PTMs dataset.
Acknowledgement
This work was supported by the Grant-in-Aid for Challenging Exploratory Research with JSPS KAKENHI Grant Number 17K20009.
References
- Knorre DG, Kudryashova NV, Godovikova TS. Chemical and functional aspects of posttranslational modification of proteins. Acta Naturae. 2009; 1: 29-51. Ref.: https://goo.gl/bHviVJ
- Xie L, Liu W, Li Q, Chen S, Xu M, et al. First succinyl-proteome profiling of extensively drug-resistant Mycobacterium tuberculosis revealed involvement of succinylation in cellular physiology. J Proteome Res. 2015; 14: 107-119. Ref.: https://goo.gl/7JwQLd
- Yang M, Yang J, Zhang Y, Zhang W. Influence of succinylation on physicochemical property of yak casein micelles. Food Chem. 2016; 190: 836-842. Ref.: https://goo.gl/eqErGv
- Rohira AD, Chen CY, Allen JR, Johnson DL. Covalent small ubiquitin-like modifier (SUMO) modification of Maf1 protein controls RNA polymerase III-dependent transcription repression. J Biol Chem. 2013; 288: 19288-19295. Ref.: https://goo.gl/WG8vq3
- Medzihradszky KF. Peptide sequence analysis. Methods Enzymol. 2005; 402: 209-244. Ref.: https://goo.gl/9Kfp94
- Agarwal KL, Kenner GW, Sheppard RC. Feline gastrin. An example of peptide sequence analysis by mass spectrometry. J Am Chem Soc. 1969; 91: 3096-3097. Ref.: https://goo.gl/tck65Z
- Welsch DJ, Nelsestuen GL. Amino-terminal alanine functions in a calcium-specific process essential for membrane binding by prothrombin fragment 1. Biochemistry. 1988; 27: 4939-4945. Ref.: https://goo.gl/FwgX1a
- Slade DJ, Subramanian V, Fuhrmann J, Thompson PR. Chemical and biological methods to detect post-translational modifications of arginine. Biopolymers. 2014; 101: 133-143. Ref.: https://goo.gl/qBW8uZ
- Umlauf D, Goto Y, Feil R. Site-specific analysis of histone methylation and acetylation. Methods Mol Biol, 2004; 287: 99-120. Ref.: https://goo.gl/zjNS6r
- Jaffrey SR, Erdjument-Bromage H, Ferris CD, Tempst P, Snyder SH. Protein S-nitrosylation: a physiological signal for neuronal nitric oxide. Nat Cell Biol. 2001; 3: 193-197. Ref.: https://goo.gl/q2hteS
- Doll S, Burlingame AL. Mass spectrometry-based detection and assignment of protein posttranslational modifications. ACS Chem Biol. 2015; 10: 63-71. Ref.: https://goo.gl/fZ5uQy
- Richards AL, Hebert AS, Ulbrich A, Bailey DJ, Coughlin EE, et al. One-hour proteome analysis in yeast. Nat Protoc. 2015; 10: 701-714. Ref.: https://goo.gl/NjFpTb
- Hebert AS, Richards AL, Bailey DJ, Ulbrich A, Coughlin EE, et al. The one hour yeast proteome. Mol Cell Proteomics. 2014; 13: 339-347. Ref.: https://goo.gl/WsZKTg
- Imamura H, Sugiyama N, Wakabayashi M, Ishihama Y. Large-scale identification of phosphorylation sites for profiling protein kinase selectivity. J Proteome Res. 2014;13: 3410-3419. Ref.: https://goo.gl/1uM654
- Masuda T, Sugiyama N, Tomita M, Ishihama Y. Microscale phosphoproteome analysis of 10,000 cells from human cancer cell lines. Anal Chem. 2011; 83: 7698-7703. Ref.: https://goo.gl/3dc9dM
- Trinidad JC, Barkan DT, Gulledge BF, Thalhammer A, Sali A, et al. Global identification and characterization of both O-GlcNAcylation and phosphorylation at the murine synapse. Mol Cell Proteomics. 2012; 11: 215-229. Ref.: https://goo.gl/ceuTj1
- Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal. 2010; 3: ra3. Ref.: https://goo.gl/L9ss6F
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, et al. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009; 325: 834-840. Ref.: https://goo.gl/Aju8io
- Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, et al. Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell. 2011; 44: 325-340. Ref.: https://goo.gl/a4ADaR
- Hendriks IA, D'Souza RC, Yang B, Verlaan-de Vries M, Mann M, et al. Uncovering global SUMOylation signaling networks in a site-specific manner. Nat Struct Mol Biol. 2014; 21: 927-936. Ref.: https://goo.gl/HZn2sq
- Syka JE, Coon JJ, Schroeder MJ, Shabanowitz J, Hunt DF. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci U S A. 2004;101: 9528-9533. Ref.: https://goo.gl/wSMjGt
- Myers SA, Daou S, Affar el B, Burlingame A. Electron transfer dissociation (ETD): the mass spectrometric breakthrough essential for O-GlcNAc protein site assignments-a study of the O-GlcNAcylated protein host cell factor C1. Proteomics. 2013; 13: 982-991. Ref.: https://goo.gl/nm45xC
- Ramstrom M, Sandberg H. Characterization of gamma-carboxylated tryptic peptides by collision-induced dissociation and electron transfer dissociation mass spectrometry. Eur J Mass Spectrom (Chichester, Eng). 2011; 17: 497-506. Ref.: https://goo.gl/XouSno
- Moremen KW, Tiemeyer M, Nairn AV. Vertebrate protein glycosylation: diversity, synthesis and function. Nat Rev Mol Cell Biol. 2012; 13: 448-462. Ref.: https://goo.gl/qxaWhh
- Han X, Yang K, Gross RW. Multi-dimensional mass spectrometry-based shotgun lipidomics and novel strategies for lipidomic analyses. Mass Spectrom Rev. 2012; 31: 134-178. Ref.: https://goo.gl/fkeRkS
- Tan M, Peng C, Anderson KA, Chhoy P, Xie Z, et al. Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 2014; 19: 605-617. Ref.: https://goo.gl/jYHNdT
- Basu A, Rose KL, Zhang J, Beavis RC, Ueberheide B, et al. Proteome-wide prediction of acetylation substrates. Proc Natl Acad Sci U S A. 2009; 106: 13785-13790. Ref.: https://goo.gl/iRi8D7
- Striebel F, Imkamp F, Sutter M, Steiner M, Mamedov A, et al. Bacterial ubiquitin-like modifier Pup is deamidated and conjugated to substrates by distinct but homologous enzymes. Nat Struct Mol Biol. 2009; 16: 647-651. Ref.: https://goo.gl/YD2Y8P
- DeMartino GN. PUPylation: something old, something new, something borrowed, something Glu. Trends Biochem Sci. 2009; 34: 155-158. Ref.: https://goo.gl/XGN8T3
- Passerini A, Punta M, Ceroni A, Rost B, Frasconi P. Identifying cysteines and histidines in transition-metal-binding sites using support vector machines and neural networks. Proteins. 2006; 65: 305-316. Ref.: https://goo.gl/BnZ38n
- Youn E, Peters B, Radivojac P, Mooney SD. Evaluation of features for catalytic residue prediction in novel folds. Protein Sci. 2007; 16: 216-226. Ref.: https://goo.gl/Xrxuto
- Sharma A, Rastogi T, Bhartiya M, Shasany AK, Khanuja SP. Type 2 diabetes mellitus: phylogenetic motifs for predicting protein functional sites. J Biosci. 2007; 32: 999-1004. Ref.: https://goo.gl/KhffLS
- Vandermarliere E, Martens L. Protein structure as a means to triage proposed PTM sites. Proteomics. 2013; 13: 1028-1035. Ref.: https://goo.gl/npNYGF
- Ren J, Wen L, Gao X, Jin C, Xue Y, et al. CSS-Palm 2.0: an updated software for palmitoylation sites prediction. Protein Eng Des Sel. 2008; 21: 639-644. Ref.: https://goo.gl/8qJhj2
- Liu Z, Cao J, Ma Q, Gao X, Ren J, et al. GPS-YNO2: computational prediction of tyrosine nitration sites in proteins. Mol Biosyst. 2011; 7: 1197-1204. Ref.: https://goo.gl/h1nSr8
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997; 25: 3389-3402. Ref.: https://goo.gl/QDHQR3
- Hasan MM, Khatun MS. Recent progress and challenges for protein pupylation sites prediction. EC Proteomics and Bioinformatics. 2017; 2.1: 36-45.
- Hasan MM, Zhou Y, Lu X, Li J, Song J, et al. Computational Identification of Protein Pupylation Sites by Using Profile-Based Composition of k-Spaced Amino Acid Pairs. PLoS One. 2015; 10: e0129635. Ref.: https://goo.gl/nENNxR
- Gobel U, Sander C, Schneider R, Valencia A. Correlated mutations and residue contacts in proteins. Proteins. 1994;18: 309-317. Ref.: https://goo.gl/7nnsc4
- Lockless SW, Ranganathan R. Evolutionarily conserved pathways of energetic connectivity in protein families. Science. 1999; 286: 295-299. Ref.: https://goo.gl/gkajNd
- Dekker JP, Fodor A, Aldrich RW, Yellen G. A perturbation-based method for calculating explicit likelihood of evolutionary co-variance in multiple sequence alignments. Bioinformatics. 2004; 20: 1565-1572. Ref.: https://goo.gl/vpaeS8
- Hasan MM, Khatun MS, Mollah MNH, Yong C, Guo D. A systematic identification of species-specific protein succinylation sites using joint element features information. Int J Nanomedicine. 2017; 12: 6303-6315. Ref.: https://goo.gl/KP5B9P
- Halperin I, Glazer DS, Wu S, Altman RB. The FEATURE framework for protein function annotation: modeling new functions, improving performance, and extending to novel applications. BMC Genomics. 2008; 9 Suppl 2: S2. Ref.: https://goo.gl/QJMzEc
- Mooney SD, Liang MH, DeConde R, Altman RB. Structural characterization of proteins using residue environments. Proteins. 2005; 61: 741-747. Ref.: https://goo.gl/okAL7j
- Amitai G, Shemesh A, Sitbon E, Shklar M, Netanely D, et al. Network analysis of protein structures identifies functional residues. J Mol Biol. 2004; 344: 1135-1146. Ref.: https://goo.gl/sTTkh1
- Rani P, Pudi V. RBNBC: Repeat Based Naive Bayes Classifier for Biological Sequences. Icdm 2008: Eighth Ieee International Conference on Data Mining, 2008; Proceedings: 989-994.
- David J. Hand KY. Idiot's Bayes: Not So Stupid after All? International Statistical Review /Revue Internationale de Statistique, 2001; 69: 385-398.
- Shao J, Xu D, Tsai SN, Wang Y, Ngai SM. Computational identification of protein methylation sites through bi-profile Bayes feature extraction. PLoS One. 2009; 4: e4920. Ref.: https://goo.gl/KPoSNi
- Zhang SW, Pan Q, Zhang HC, Shao ZC, Shi JY. Prediction of protein homo-oligomer types by pseudo amino acid composition: Approached with an improved feature extraction and Naive Bayes Feature Fusion. Amino Acids. 2006; 30: 461-468. Ref.: https://goo.gl/o9AG12
- Sheppard S, Lawson ND, Zhu LJ. Accurate identification of polyadenylation sites from 3' end deep sequencing using a naive Bayes classifier. Bioinformatics. 2013; 29: 2564-2571. Ref.: https://goo.gl/tNVeZn
- Yang P, Humphrey SJ, Fazakerley DJ, Prior MJ, Yang G, et al. Re-fraction: a machine learning approach for deterministic identification of protein homologues and splice variants in large-scale MS-based proteomics. J Proteome Res. 2012; 11: 3035-3045. Ref.: https://goo.gl/MyCAHJ
- Simon P. Too Big to Ignore: The Business Case for Big Data. Wiley, 2013; 89.
- Breiman L. Random Forests. Machine Learning, 2001; 45: 5-32. Ref.: https://goo.gl/9rqw7o
- Maclin R, Opitz D. Popular ensemble methods: An empirical study. Journal of Artificial Intelligence Research. 1999; 11: 169-198. Ref.: https://goo.gl/ugm7T4
- Polikar R. Ensemble based systems in decision making. Circuits and systems magazine, IEEE. 2006; 6: 21-45. Ref.: https://goo.gl/GAnEij
- Rokach L. Ensemble-based classifiers. Artificial Intelligence Review. 2010; 33: 1-39. Ref.: https://goo.gl/naMCA5
- Brown G, Wyatt J, Harris R, Yao X. Diversity creation methods: a survey and categorisation. Information Fusion. 2005; 6: 5-20. Ref.: https://goo.gl/ABKNwa
- Adeva JJG, Beresi U, Calvo R. Accuracy and diversity in ensembles of text categorisers. CLEI Electronic Journal. 2005; 9: 1-12. Ref.: https://goo.gl/c3vzuR
- Liu ZP, Wu LY, Wang Y, Zhang XS, Chen L. Prediction of protein-RNA binding sites by a random forest method with combined features. Bioinformatics. 2010; 26: 1616-1622. Ref.: https://goo.gl/TQHQRE
- Kumar KK, Pugalenthi G, Suganthan PN. DNA-Prot: identification of DNA binding proteins from protein sequence information using random forest. J Biomol Struct Dyn. 2009; 26: 679-686. Ref.: https://goo.gl/gXLBHT
- Qi Y, Klein-Seetharaman J, Bar-Joseph Z. Random forest similarity for protein-protein interaction prediction from multiple sources. Pac Symp Biocomput. 2005; 531-542. Ref.: https://goo.gl/kU7VD1
- Hasan MM, Guo D, Kurata H. Computational identification of protein S-sulfenylation sites by incorporating the multiple sequence features information. Mol Biosyst. 2017; 13: 2545-2550. Ref.: https://goo.gl/JhMKEE
- Hasan MM, Yang S, Zhou Y, Mollah MN SuccinSite: a computational tool for the prediction of protein succinylation sites by exploiting the amino acid patterns and properties. Mol Biosyst, 2016; 12: 786-795. Ref.: https://goo.gl/Zezfm1
- Cornia C, Vapnik V. Support-vector networks. Machine Learning. 1995; 20: 273-297. Ref.: https://goo.gl/RE4bJo
- Chang CC. LIBSVM: A Library for Support Vector Machines. ACM transactions on intelligent systems and technology. 2011; 2. Ref.: https://goo.gl/Jx29pP
- Pavlidis P, Wapinski I, Noble WS. Support vector machine classification on the web. Bioinformatics. 2004; 20: 586-587. Ref.: https://goo.gl/guqAUu
- Frank E, Hall M, Trigg L, Holmes G, Witten IH. Data mining in bioinformatics using Weka. Bioinformatics. 2004; 20: 2479-2481. Ref.: https://goo.gl/QQdQtq
- Chen X, Qiu JD, Shi SP, Suo SB, Liang RP. Systematic analysis and prediction of pupylation sites in prokaryotic proteins. PLoS One. 2013; 8: e74002. Ref.: https://goo.gl/h8t9mH
- Tung CW. Prediction of pupylation sites using the composition of k-spaced amino acid pairs. J Theor Biol. 2013; 336: 11-17. Ref.: https://goo.gl/AhZmz8
- Wu S, Zhang Y. A comprehensive assessment of sequence-based and template-based methods for protein contact prediction. Bioinformatics. 2008; 24: 924-931. Ref.: https://goo.gl/BsZmRP
- Yan RX, Si JN, Wang C, Zhang Z. DescFold: a web server for protein fold recognition. BMC Bioinformatics. 2009; 10: 416. Ref.: https://goo.gl/NaWMFM
- Guo J, Chen H, Sun Z, Lin Y. A novel method for protein secondary structure prediction using dual-layer SVM and profiles. Proteins. 2004; 54: 738-743. Ref.: https://goo.gl/hNVe7r
- Minsky MSP. An Introduction to Computational Geometry. 1969; ISBN 0-262-63022-2.
- Fukushima K. Cognitron: a self-organizing multilayered neural network. Biol Cybern, 1975; 20: 121-136. Ref.: https://goo.gl/hzsy1e
- Tang YR, Chen YZ, Canchaya CA, Zhang Z. GANNPhos: a new phosphorylation site predictor based on a genetic algorithm integrated neural network. Protein Eng Des Sel. 2007; 20: 405-412. Ref.: https://goo.gl/GJH3G8
- Blom N, Sicheritz-Ponten T, Gupta R, Gammeltoft S, Brunak S. Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics. 2004; 4: 1633-1649. Ref.: https://goo.gl/dGmYaQ
- Dehouck Y, Grosfils A, Folch B, Gilis D, Bogaerts P, et al. Fast and accurate predictions of protein stability changes upon mutations using statistical potentials and neural networks: PoPMuSiC-2.0. Bioinformatics. 2009; 25: 2537-2543. Ref.: https://goo.gl/BhKBfr
- Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999; 292: 195-202. Ref.: https://goo.gl/nUkouC
- McGuffin LJ, Bryson K, Jones DT. The PSIPRED protein structure prediction server. Bioinformatics. 2000; 16: 404-405. Ref.: https://goo.gl/UW6fu4
- Bienkowska JR, Dalgin GS, Batliwalla F, Allaire N, Roubenoff R, et al. Convergent Random Forest predictor: methodology for predicting drug response from genome-scale data applied to anti-TNF response. Genomics. 2009; 94: 423-432. Ref.: https://goo.gl/55hyK