The bio-energy transport in the protein molecules and its experimental validations of correctness

Pang Xiao-Feng*
Institute of Life Science and Technology, University of Electronic Science and Technology of Chengdu, China
*Address for Correspondence: Pang Xiao-Feng, Institute of Life Science and Technology, University of Electronic Science and Technology of Chengdu 610054, China; Tel: +86 28 8320 2316; E-mail: [email protected]
Dates: Submitted: 29 December 2017; Approved: 15 January 2018; Published: 18 January 2018
How to cite this article: Pang XF. The bio-energy transport in the protein molecules and its experimental validations of correctness. Ann Proteom Bioinform. 2018; 2: 001-048. DOI: 10.29328/journal.apb.1001004
Copyright License: © 2018 Pang XF. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Bio-energy transport; Solution; Experimental evidence, Infrared spectrum; Raman spectrum; Protein; Acetanilide; Collagen; E.Coli., Lifetime; Measurement; Specific heat

Abstract
The bio-energy released by the hydrolysis of adenosine triphosphate, which relate to plenty of life activities and is transported in a solution, and its theory of transport are first stated and built in helix protein molecule. In order to confirm and verify the correctness of the transported theory we here systematically summarized and reviewed a great number of experimental investigation and evidences obtained by us and other researchers in past 30 years, involving the real existences of the solution and its features and lifetimes. In this survey we outlined and presented concretely the features of infrared spectra of absorption, Raman spectra and specific heat of the molecular crystal-acetanilide collagen, bivine serum albumin, myoglobin proteins and E.Coli. cell as well as the lifetimes of the solution in acetanilide and myoglobin measured by using pump-probe techniques and free-electron laser experiment, in which we give not only experimental data but also their comparisons with theoretical results. These experimental data and evidences provided here are enough to verify and affirm the true existences of the new solution, which can complete itself functions of bio-energy transport in the lifetime, and the correctness of the new theory of bio-energy transport in the acetanilide and protein molecule. Thus we can affirm the correctness of theory of the bio-energy transport in helix protein molecule, which can greatly promote the development of molecular biology.
Introduction
The biological energy and its theory of transport
The so-called life is just processes of mutual changes and coordination of the bio-material, bio-energy and bio-information, their synthetic movements and cooperative and coordinate changes each other are just total life activity in the live systems in the light of biophysicist’s view, in which the bio-material is the foundation if life, the bio-energy is its center, the bio-information is the key of life activity, but the transformation and transfer of bio-information are always accompanied with the transport of bio-energy in living systems [1]. Therefore, the bio-energy and its transport are the fundamental and an important process in life activity. As a matter of fact, many biological processes, such as muscle contraction, DNA reduplication, neuroelectric pulse transfer on the neurolemma and work of calcium pump and sodium pump in cells, and so on, are associated always with bio-energy transport in the life bodies, where the bio-energy comes mainly from the reaction of hydrolysis of adenosine triphosphate (ATP), in which the energy released in the reaction of an ATP molecule with water is 0.43eV under normal physiological conditions. The reaction can be represented by
Where ADP is the adenosine diphosphate. Just so, there are always a biological process of energy transport from production places to absorption sites in the living systems. Then investigation of bio-energy transport in bio-tissues and determination of its rules have very important significance in life science. However, understanding the mechanism of the transport in living systems has been a long-standing problem that remains of great interest up to now in life science.
Davydov’s model of bio-energy transport and its corrections
In general, the bio-energy is transported along the protein molecules, in which the energy can be converted to a particular vibrational excitation within the protein molecule. A likely recipient exchange is the amide-I vibration. Their vibration is primarily a stretch and contraction of the C=O bond of the peptide groups (or amino acid residues). The amide-I vibration is also a prominent feature in infrared and Raman spectra of protein molecules. Experimental measurement shows that one of the fundamental frequencies of the amide-I vibration is about 0.205eV. This energy is about half the energy released during the ATP hydrolysis. Moreover, it remains nearly constant from protein to protein, which indicates that it is rather weakly coupled to other degrees of freedom. All these factors lead to the assumption that the energy released by ATP hydrolysis might stay localized and stored in the amide-I vibration excitation. As an alternative to electronic mechanisms [2-4], one can assume that the energy is stored as vibrational energy of the C=0 stretching mode (amide-I) in a protein polypeptide chain. Following Davydov’s idea [5-12], ones take into account the coupling between the amide-I vibrarional quantum (exciton ) and the acoustic phonon (molecular displacements) in the amino acid residues, through the coupling, nonlinear interaction appears in the motion of the vibrartional quanta, which could lead to a self-trapped state of the vibrational quantum. The latter plus the deformational amino acid lattice together can travel over macroscopic distances along the molecular chains, retaining the wave shape, energy, momentum and other properties of the quasiparticle. In this way, the bio-energy can be transported as a localized “wave packet” or solution. This is just the Davydov’s model of bio-energy transport in proteins, which was proposed in the 1970s [5-7].
Davydov’s model of bio-energy transport worked athelical proteins as shown in figure 1. Following Davydov’s idea [5-14], the Hamiltonian describing such system is of the form
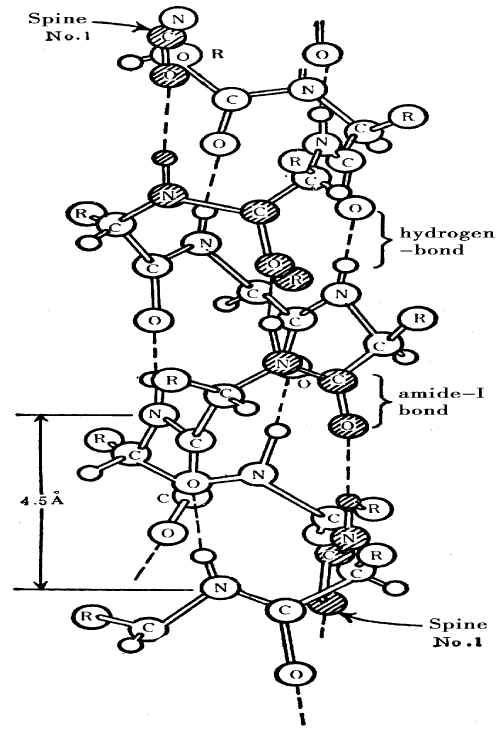
Figure 1: Molecular structure of α- helix protein molecule.
In Davydov’s model, where
=0.205ev is the amide-I quantum energy, -J is the dipole-dipole interaction energy between neighbouring amides,
is the creation (annihilation) operator for an amide-I quantum (exciton) in the site n, un is the displacement operator of lattice oscillator at site n, Pn is its conjugate momentum operator, M is the mass of an amino acid residue, w is the elasticity constant of the protein molecular chains, and χ1 is an nonlinear coupling parameter and represents the coupling size of the exciton- phonon interaction in the protein molecules. The wave function of states of the systems in Davydov model is of the form [5-14]:
where I0>=I0>ex Io>ph, I0>ex and I0>ph are the ground states of the exciton and phonon, respectively,
is annihilation (creation) operator of the phonon with ware vector q,
and
and
are Some undetermined functions of time. The Davydov’s solution obtained from Equations (1)-(2) in the semiclassical limit and using the continuum approximation has the form of
Which corresponds to an excitation localized over a scale r0/µD, where
is the sound speed in the protein molecular chains, v is the velocity of the solution, r0 is the lattice constant. Evidently, the solution contains only one exciton because N=<
. This shows that the Davydov’s solution is formed through self-trapping of one exciton, its binding energy is
Davydov’s idea yields a compelling picture for the mechanism of bioenergy transport in protein molecules and consequently has been the subject of a large number of works [15-103]. A lot of issues related to the Davydov;s model, including the foundation and accuracy of the theory, the quantum and classical properties and the thermal stability and lifetimes of the Davydov solution have extensively been studied by many scientists [36-49]. However, considerable controversy has arisen concerning whether the Davydov’s solution is sufficiently stable in the region of biological temperature to provide a viable explanation for bio-energy transport. It is out of question that the quantum fluctuations and thermal perturbations arising from the temperature of the systems are expected to cause the Davydov’s solution to decay into a delocalized state. Some numerical simulations indicated that the Davydov’s solution is not stable at the biological temperature 300K [36-49]. Other simulations showed that the Davydov’s solution is stable at 300K [36-49], but they were based on classical equations of motion which are likely to yield unreliable estimates for the stability of the solution [2-4]. The simulations based on the ID2> state in Equation (2) generally show that the stability of the solution decreases with increasing temperatures and that the solution is not sufficiently stable in the region of biological temperature. Since the dynamical equations used in the simulations are not equivalent to the nonlinear Schrodinger equation, the stability of the solution obtained by these numerical simulations is unavailable or unreliable. The simulation [9] based on the ID1>state in Equation (3) with the thermal treatment of Davydov [15-20], where the equations of motion are derived from a thermally averaged Hamiltonian, yields the confusing result that the stability of the solution is enhanced with increasing temperature, predicting that ID1>-type solution is stable in the region of biological temperature. Evidently, the conclusion is doubtful because the Davydov procedure in which an equation of motion for an average dynamical state obtained from an average Hamiltonian, corresponding to the Hamiltonian averaged over a thermal distribution of phonons, is inconsistent with standard concepts of quantum-statistical mechanics in which a density matrix must be used to describe the system. Therefore, any fully exact quantum- mechanical treatment for the numerical simulation of the Davydov’s solution does not exist. However, for the thermal equilibrium properties of the Davydov solution, there is a quantum Monte Carlo simulation [64,65]. In this study the correlation characteristic of solutionlike quasiparticles occur only at low temperatures, about T<10k for widely accepted parameter values of protein molecules. This is consistent at a qualitative level with the result of Cottingham et al. [66,67]. The latter is a straightforward quantum- mechanical perturbation calculation, the lifetime of the Davydov’s solution obtained by using this method is too small (about 10-12 – 10-13 sec) to be useful in biological processes. This indicates clearly that the Davydov solution is not a true wave function of the systems. A through study in terms of parameter values, different types of nonuniformity, different thermalization schemes, different wave functions, and different associated dynamics leads to a very complicated picture for the Davydov’s model [44-63]. These results do not completely rule out the Davydov’s theory, however they do not eliminate the possibility of another wave function and a more sophisticated Hamiltonian of the system having a solution with longer lifetimes and good thermal stability.
Indeed, the question of the lifetime of the solution in protein molecules is twofold. In Langevin dynamics, the problem consists of uncontrolled effects arising from the semiclassical approximation. In quantum treatments, the problem has been the lack of an exact wave function for the solution. The exact wave function of the fully quantum Davydov’s model has not been known up to now. Different wave functions have been used to describe the states of the fully quantum-mechanical systems [21-32]. Although some of these wave functions lead to exact quantum states and exact quantum dynamics in the J=0 state, they also share a problem with the original Davydov’s wave function, namely that the degree of approximation included when
is not known. Therefore, it is necessary to reform Davydov’s wave function. Scientists had though that the solution with a multiquantum (), for example, the coherent state of Brown et al. [21-28], the multiquantum state of Kerr et al. [62,63], and Schweitzer et al. [66,67], the two- quantum state of Cruzeiro-Hansson [44-49] and Forner [74], and so on, would be thermally stable in the region of biological temperature and could provide a realistic mechanism for bio-energy transport in protein molecules. However, the assumption of the standard coherent state is unsuitable or impossible for biological protein molecules because there are innumerable particles in this state and one could not retain conservation of the number of particles of the system. The assumption of a multiquantum state (n>2) along with a coherent state is also inconsistent with the fact that the bioenergy released in ATP hydrolysis can excite only two quanta of amide-I vibration. Although the numerical result shows that the solution of two-quantum state is more stable than that with a one-quantum state, the models have many problems and are not suitable to proteins [104].
Pang’s model of bio-energy transport and its features
From serious study of Davydov’s model we can find that it is indeed too simple, i.e., it does not denote the elementary properties of the collective excitations occurring in protein molecules, and many improvements of it have also been unsuccessful. In fact, Davydov operation is not strictly correct. A basic reason for the failure of the Davydov model is just that it ignores completely the above important properties of the protein molecules. Let us consider the Davydov model in the present viewpoint. First, as far as the Davydov wave functions, both D1 > and D1, are concerned, they are not true solutions of the protein molecules. On the one hand, there is obviously asymmetry in the Davydov wave function since the phononic parts is a coherent state, while the excitonic part is only an excitation state of a single particle. It is not reasonable that the same nonlinear interaction generated by the coupling between the excitons and phonons produces different states for the phonon and exciton. Thus, Davydov’s wave function should be modified [72-103], i.e., the excitonic part in it should also be coherent or quasicoherent to represent the coherent feature of collective excitation in protein molecules. However, the standard coherent [21-28] and large-n excitation states [62,63] are not appropriate to the protein molecules due to the reasons mentioned above. Similarly, Forner’s and Cruzeiro-Hansson’s two-quantum states do not fulfill the above request.
On the basis of the work of Cruzeio-Hansson, Forner, Schweitzer and Takeno and Pang, both the Hamiltonian and the wave function of the Davydov model [10-40] have been improved and developed by Pang [104-126], in which Davydov’s wave function has been replaced with a quasi-coherent two-quanta state for exhibiting the coherent behaviors of collective excitations [127-130] which are a feature of the energy released in ATP hydrolysis in the systems. The new wave function is represented [104-126] by
Where
and
are boson creation and annihilation operators for the exciton
,
and
are the ground states of the exciton and phonon, respectively un and Pn are the displacement and momentum operators of the lattice oscillator at site n, respectively. The
,
and
are three sets of unknown functions, λ is a normalization constant. It is assumed hereafter that λ=1 for convenience of calculation, except when explicitly mentioned.
The second problem arises from the Davydov Hamiltonian [5-14]. Now that the Davydov’s Hamiltonian takes into account the resonant or dipole-dipole interaction of the neighboring amide-I vibrational quanta in neighboring peptide groups with an electrical moment of about 3.5D, but why do we consider not the changes of relative displacement of the neighboring peptide groups arising from this interaction? This is obviously not reasonable. This means that we should add the new interaction term
into the Davydov’s Hamiltonian for representing the correlations of the collective excitations and collective motions in the protein molecules [76-103]. Although the dipole- dipole interaction is small as compared with the energy of the amide-I vibrational quantum, the change of relative displacement of neighboring peptide groups resulting from this interaction cannot be ignored due to the sensitive dependence of the dipole-dipole interaction on the distance between amino acids in the protein molecules i9n the soft condensed matter and bio-self-organization. Thus, the Davydov’s Hamiltonian is replaced now by
Where
= 0.205eV is the energy of the exciton (the C=0 stretching mode). The present nonlinear coupling constants are
, which represent the modulations of the on-site energy and resonant (or dipole-dipole) interaction energy of excitons caused by the molecules displacements, respectively .M is the mass of an amino acid molecule and w is the elasticity constant of the protein molecular chains. J is the dipole-dipole interaction energy between neighboring sites. The physical meaning of the other quantities in Equation (7) are the same as those in the above explanations.
The new Hamiltonian and wave function in Pang’s model shown in Equations (6)-(7) are different from Davydov’s. We add a new interaction term
, into the original Davydov Hamiltonian. Thus the Hamiltonian now has better one-by –one correspondence of the interactions and can represent the features of mutual correlations of the collective excitations and of collective motions in the protein molecules. However, we here should point out that the different coupling between the relevant modes was also considered by Takeno et al. [75-77,131,132] and Pang [78-103] in the Hamiltonian of the vibron-solution model for one-dimensional oscillator-lattice and protein systems, respectively, but the wave functions of the systems they used are different from Equations (6)-(7).
Obviously, the present wave function of the exciton in Equation (6) is not an excitation state of single particle, but rather a coherent state, more precisely, a quasicoherent state, because it retain only for three terms of the expansion of a standard coherent state,. Which thus can be viewed as an effective truncation of a standard coherent state. It is clear that when small
, i.e.
, we can represent the wave function of the excitons
, in Equation (6)[104-126] by
The last representation in Equation (8) is a standard coherent state. Therefore, the state of exciton denoted by the wave function
has a coherent feature. In the meanwhile, we can verify that the new wave function in Equation (6) is also normalized at λ = 1. Since the condition of
is required in the calculation, then the above condition of
also is naturally satisfied for the protein molecules consisting of several hundreds or thousands of amino acid residues. Thus the above representation in Equation (8) is justified and correct for the protein molecules. Since the coherent state is certainly normalized, then the wave function
in Equation (6), which can be represented as a standard coherent state, is exactly normalized at λ=1. Clearly, the above demonstration for the normalization is correct, reasonable and credible because it is obtained from a strict mathematical, physical and biological theory. Thus we have not any reasons to doubt the normalization of wave function in Equation (6). However, it is not an eigenstate of the number operator because of
However, in this state the numbers of quanta are determinate instead of innumerable. Since the third Term in the exciton part in Pang’s wave function contains the coefficient of “1/2!”, which guarantees that the third term in the exciton’s wave function contribute only one quantum, then we find that the state contains number of exciton by computing the expectation value of the number operator
in this state and sum over the states, i.e.,
Therefore,
> contains only two quanta, instead of one quantum or three quanta, i.e., it represents exactly a coherent superposition of the excitonic state with two quanta and the ground state of the exciton. Thus the new wave function not only exhibits the coherent feature of the collective excitation of excitons and phonons caused by the nonlinear interaction generated by virtue of the exciton-phonon interaction, which makes the wave function of the states of the system symmetrical, but also agrees with the fact that the energy released in the ATP hydrolysis (about 0.43 eV) can create two amide-I vibrational quanta (exciton), which, then, can also make the numbers of excitons maintain conservation in the Hamiltonian, Equation (7). Thus it is correct and available. We here refer to it as a two quanta quasicoherent state. Obviously, it is completely different from Davydov’s, which is an excitation state of a single particle with one quantum and an eigenstate of the number operator. At the same time, the new wave function in Equation (6) is either two-quantum states proposed by Forner [74] and Cruzeiro-Hansson [44-49] or a standard coherent state proposed by Brown et al. [21-28], and Kerr et al. [62-63], and Schweitzer et al. [66-67], multiquanta states. Therefore, the wave function, Equation (6), is new for the protein molecular systems. In the meanwhile, the new wave function in Pang’s model has the following advantages, i.e., the equation of motion of the solution in the system can also be obtained from the Heisenberg equations of the creation and annihilation operators in quantum mechanics using Equations (6) and (7). However, it is impossible for the wave function of state of the system in other models, including the one-quantum state [5-14] and the two-quantum state [44-49]. Therefore, the above Hamitonian and wave function, Equations (6) and (7), are both new and appropriate to the protein molecules.
We know from Equation (6) that the phonon part depending on the displacement and momentum operators in the new wave function in Equation (6) is a coherent state of the normal model creation and annihilation operators. A coherent state for the mode with wave vector q is denoted by
[5-14,62,63,104-124]. Utilizing again the standard transformations:
We can get
, where
is same in Equation (6) and
[62,63], r0 Is the distance between neighboring amino acid molecules, and
is the annihilation (Creation) operator of the phonon with wave vector q. utilizing the above results and the formulas of the expectation values of the Heisenberg equations of operators, un and Pn, in the state
.
,
,
,
and
the time-dependent Shrodinger equation [79-105],
, in the continuum approximation we get we can obtain:
and
here
and
,
The solution solution of Equation (11) [131-133] is thus
with
,
,
The above treatment yields a localized coherent structure with size of order 2π r0/μp that propagates with velocity v and can transfer energy ES01<
. Unlike bare excitons that are scattered by the interactions with the phonons, this solution state describes a quasi-particle consisting of the two excitons plus a lattice deformation and hence a priori includes interaction with the acoustic phonons. So the solution is not scattered and can spread through maintaining its form, energy, momentum and other quasiparticle properties after moving over a macroscopic distance. The bell-shaped form of the solution in Equation (13) also does not depend on the excitation method. Hence, it is self-consistent. Since the solution always move with velocity v less than that of longitudinal sound v0 in the chains then they do not emit phonons, i.e., its kinetic energy is not transformed into thermal energy. This just the important reason for the high stability of the new solution. In addition, the energy of the solution state is below the bottom of the bare exciton bands, the energy gap between them being
for small velocity of propagation. Hence there is an energy penalty associated with the destruction with transformation from the solution state to a bare exciton state, i.e, the destruction of the solution state requires simultaneous removal of the lattice distortion. We know that the transition probability to a lattice state without distortion is very small, in general, being negligible for a long chain. Considering this it is reasonable to assume that such a solution is stable enough to propagate through the length of a typical protein structure. However, the thermal stability of the solution state must be calculated quantitatively. The following calculation addresses this point explicitly.
Although the forms of the above equations of motion and the corresponding solution, Equations (11)-(13), are quite similar to those of the Davydov solution, the properties of the new solution have very large differences from the latter because the parameter values in the equation of motion and the solution Equations (11) and (13), including R(t) Gp, and up, have obvious distinctions from those in the Davydov model. A straightforward result of the new model is to increase the nonlinear interaction energy Gp(Gp= 2GD
and the amplitude of the new solution, and decrease its width due to an increase of
when compared with Davydov solution, where
and
are the corresponding values in Davydov’s model. Thus the localized feature of the new solution is enhanced. Therefore, its stability against the quantum fluctuation and thermal perturbations is increased considerably as compared with the Davydov’s solution.
The energy of the new solution in Equation (13) in the improved model can be represented [131,132] by
The rest energy of the new solution in Pang;s model is
where
is
the energy of deformation of the amono acid lattice. The effective mass of the new solution is
. In such a case, the binding energy of the new solution is
Namely, EBP is larger than that of the Davydov solution. The latter is . We can estimate that the binding energy of the new solution is about several decades larger than that of the Davydov solution. This is a very interesting result. It is helpful to enhance thermal stability of the new solution. Obviously, the increase of the binding energy of the new solution comes from its two-quantum nature and the added interaction
, in the Hamiltonian of the systems, Equation (7). However, we see from Equation (16) that the former plays the main role in the increase of the binding energy and the enhancement of thermal stability for the new solution relative to the latter due to
. The increase of binding energy results in a significant change of property of the new solution, which are discussed as follows. In comparing various correlations to this model, it is helpful to consider them as a function of a composite coupling parameter like that of Young et al. [133] and Scott [15-20] that can be written as
,
where
is the band edge for acoustic phonons (or Debye frequency). If
, it is said to be weak. Using widely accepted values for the physical parameters in the alpha helix protein molecule [5-126]:
,
We can estimate that the coupled constant lies in the region of
but 4παD = 0.036 - 0.045 for the Davydov’s model. Hence, the new model is not a weakly coupled theory as compared with the Davydov’s model. Using again the notation of Venzel and Fischer [134], Nagy [135] and Wagner and Kongeter [136], it is convenient to define another composite parameter [15-20]:
. In terms of the two composite parameters, 4παp and γ, the solution binding energy in the new model can be written by
,
From the above parameter values in Equation (18), we obtain
. Utilizing these values, the EBP/J versus
relations in Equation (18) are plotted in figure 2. However,
for the Davydov model, where
and)
, then the EBD/J versus
relation is also plotted in figure 2. From this figure we see that the difference of solution binding energies between two models becomes larger with increasing
.
Meanwhile, we see clearly from Equations (12)-(15) and (16) that the localized feature of the new solution is enhanced due to increases of the nonlinear interaction and its binding energy resulting from the increases of exciton-phonon interaction in the improves model. Thus, the stability of the new solution in Pang’s model against quantum and thermal fluctuations is enhanced considerately. In fact, the nonlinear interaction energy forming the siliton in Pang’s model is
,
, and it is larger than the linear dispersion energy,, i.e., the nonlinear interaction in Pang’s model is so large that it can actually cancel or suppress the dispersion effect in the equation of motion ,thus the new solution is stable in such a case according to the solution theory [5-14,137].
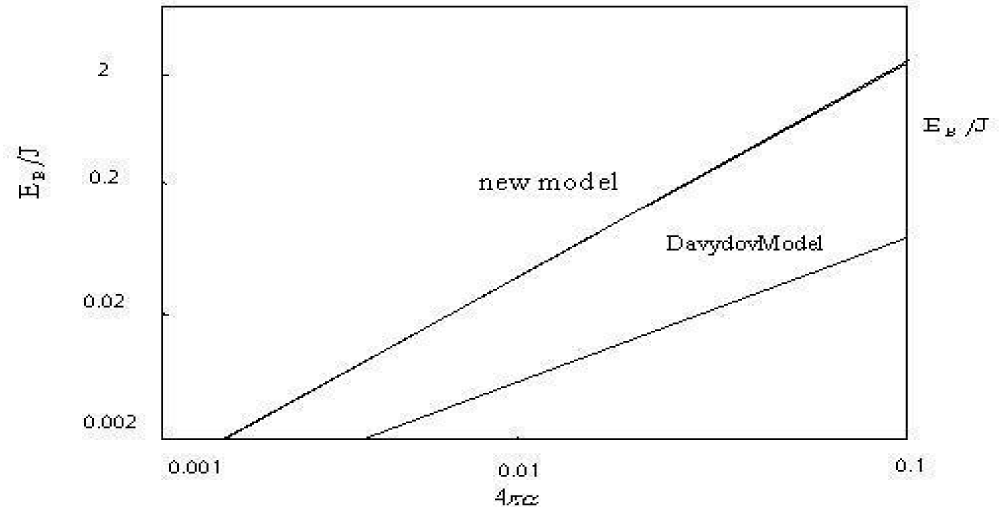
Figure 2: The binding energy (EB) of the solitons in units of dipole-dipole interaction energy (J) vs the coupled constant, 4πα , relation in Pang’s model and the Dvydovy’s model.
On the other hand, the nonlinear interaction energy in the Davydov model is
, and it is about three to four times smaller than Gp. Therefore, the stability of the Davydov’s solution is weaker as compared with the new solution. Moreover, the binding energy of the new solution in Pang’s model is
in Equation (15), which is somewhat larger than the thermal perturbation energy,
, at 300K and about four times larger than the Debye energy
,(there
is the Debye frequency). This shows that transition of the new solution to a delocalized state by the heat energy can be suppressed by the large energy difference between the initial (solution) state and final (delocalized) state, which is very difficult to compensate for with the energy of the absorbed phonon. Thus, the new solution is robust against quantum and thermal fluctuations, therefore it has a large lifetime and good thermal stability in the region of biological temperature. In practice, according to Schweitzer et al., studies (i.e the lifetime of the solution increases as
and
increase at a given temperature) [67,68] and from the above results obtained we inferred that the lifetime of the new solution will increase considerably relative to that of the Davydov solution due to the increases of
and
because the latter are about three times larger than that of the Davydov model.
On the other hand, the binding energy of Davydov solution,
is about 23 times smaller than that of the new solution, about 22 times smaller than KBT and about 6 times smaller than
, respectively. Therefore, the Davydov solution is easily destructed by the thermal perturbation energy and quantum fluctuations. Thus we can naturally infer that the Davdov solution has only a very small lifetime, and is unstable at the biological temperature 300K. This conclusion is consistent at a qualitative level with the results of Wang et al. [64,65] and Cottingham et al. [66-67].
In the above investigation of the influences of quantum and thermal effects on solution state, which are expected to cause the solution to decay into delocalized states, we postulate that the model Hamiltonian and the wave function in Pang’s model together give a complete and realistic picture of the interaction properties and allowed states for the protein molecules. The additional interaction term added in the Hamiltonian gives a one-to-one correspondence of interactions in the new model. Thus we can affirm that the new wave function in Pang’s model is a reasonable choice for the protein molecules because it not only can exhibit the coherent features of collective excitations arising from the nonlinear interaction between the exactions and phonons, but also retain the conservation of number of particles and satisfy further the fact that the energy released by the ATP hydrolysis can only excite two quanta. In such a case, using a standard calculating method [2,26] and widely accepted parameters in Equation (17) we can obtain the region encompassed of the excitation or the linear extent of the new solution,
, which is greater than the lattice constant r0 i.e.,
. Meanwhile, we can explicitly calculate the amplitude squared of the new solution using Equation (13) in its rest frame, which is
. Then the probability to find the new solution outside a range of width r0 is about 0.10. This means that the new solution is very well localized in this condition. Meanwhile, this extent of the new solution we obtained meets
, which means that the quasi-coherent solution can spread over more than one lattice spacing in the system. Thus, this proves that assuming of the continuous approximation used in the calculation is valid because the solution widths is large than the order of the lattice spacing.
Finally we calculate fully the values of the main parameters in the new model by the above values. These values and the corresponding values in the Davydov model are simultaneously listed in table 1. From table 1 we can see clearly that Pang’s model produces considerable changes in the properties of the solution, for example, large increase of the nonlinear interaction, binding energy and amplitude of the new solution, and decrease of its width as compared to those of the Davydov solution. This shows that the solution in Pang’s model is more localized and more robust against quantum and thermal fluctuations, thus its stability is enhanced [5-14,104-126]. This implies an increase in lifetime for the new solution. From Equation (16) we also find that the effect of the two-quantum nature is larger than that of the added interaction. We thus can refer to the new solution as quasi-coherent two-quantum state, which is novel and correct wave function.
| Table 1: Comparison of parameters used in the Davydov model and Pang’s model. |
Parameters
Models |
|
G(×10-21J) |
Amplitude of solution A’ |
Width of solution (×10-10m) |
Binding energy of solution EB
(×10-21J) |
Pang’s
Model |
5.94 |
3.8 |
1.72 |
4.95 |
-4.3 |
Davydov
model |
1.90 |
1.18 |
0.974 |
14.88 |
-0.188 |
The necessity of experimental verification of validity for the theoretical models
We exhibit the properties, successful and problems of different models in the above investigation [100-126], which but leaves behind many questions that are worth to study continuously. Actually, the above results were obtained analytically, based on many hypotheses, in which the protein molecules, which were used by the researchers, was regarded as a periodically and uniformly infinite-long chains composed of amino acid residues with same weight. Obviously, these features of the proteins are not completely conformable with the biological protein molecules in the living systems. As it is known, the biological proteins are an finite –long structure, which are composed of several hundreds or thousands amino acid residues with different molecular weights between 75 mp (glycine) and 204 mp (tryptophan), which correspond to variations in mass between
, where
=114mp is the average mass of an amino acid residue and mp is the proton mass. And the biological proteins adhere often a lot of side groups. Clearly, these structures affect necessarily the dynamic features of protein molecules. This means that there are a structure nonuniformity and disorder in biological-protein molecules, which result necessarily in the fluctuations of the spring constant, the dipole-dipole interaction, the exciton-phonon coupling constant and the diagonal disorder in dynamic equations in protein molecules mentioned above. Thus, the states of the solutions obtained from the theoretical models will be changed under influence of these structure disorders in the biological protein molecules. Otherwise, in the above investigation all physical parameters of the protein molecules were represented by their average values, and some approximation methods, such as long- wavelength approximation, continuum approximation, or long-time approximation, were also used in concrete calculations, which cannot be evidently used in the biological proteins. Careri et al. [137-142], demonstrated that even relatively small amounts of disorder in an amorphous film of acetanilide (ACN), a protein-like crystal, is enough to destroy the spectral signature of a “solution”. Therefore, we have the reasons to doubt the real existence of the solutions and the correctness of the above theory of bio-energy transport in protein molecules.
At the same time, the biological proteins work always at physiological temperature and biological solution containing water molecules and other ions. The thermal perturbation and damping of medium arising from the temperature will also change the states and lifetimes of the solutions, which are the carrier of the bio-energy transport. These water molecules and other ions existed in the solution will also influence the dynamic features of the protein molecules to serve as some imported impurities. On the other hand, in the α-helix protein molecules, which are constructed by three channels, there are also chain-chain interaction among the three channels, which will also disperse the energy of the solution, then the states of the solutions will be influenced. In such a case, we have the reason to doubt the validity of the above theories of bio-energy transport. Then it is very necessary to verify whether these models are appropriate to biological proteins? Whether can the models represent the real features of biological protein molecules? These problems need be eventually checked and assessed by the experimental results. Therefore, the experimental evidences play a key role in the judgment of validity of the theory of bio-energy transport. Just so, many scientists researched/are researching these problems using different methods and bio-samples, and obtained a lot of interesting results, which can be used to verify the correctness of the theory. In the following we will describe, summarize and review systematically and completely these experimental results obtained by many scientists in the area.
Experimental validation in Infrared and Raman spectra in molecular crystal- acetanilide
Molecular structure of acetanilide: The first experimental evidence for the existence of solution state mentioned above was obtained from the crystal of acetanilide (ACN)
[137-142]. ACN is composed of two close chains of hydrogen-bonded amide-I groups which consists of atoms of carbon, oxygen, nitrogen and hydrogen (CONH), in which the two chains run through the acetanilide crystal, hence, it is called a protein-like crystal [137-142]. Thus the experimental validations of the theory of bio-energy transport focus all ACN. Its crystal structure has been determined and a unit cell of acetanilide is shown in figure 3. Its space group is
and the unit cell or factor group is
for this crystal. The average lattice constants are a = 1.9640 nm, b = 0.9483 nm, and c = 0.7979 nm. There are eight molecules in a unit cell and at the amide- I frequency, each of these has one degree of freedom (d.f.). Thus, there are three infrared-active modes (
), four Raman-active modes (
), and one inactive mode (
). However, at low frequency (<200
), each molecule exhibits 6 d.f. (three translations and three rotations). This gives 48 low-frequency modes: 24 Raman active modes (
), 18 infrared-active modes (
) and six (
) modes corresponding to acoustic modes of translation and rotation). All of these active modes are seen in infrared absorption and Raman experiments. Meanwhile, in acetanilide, the nearly planar amide-I groups have bond lengths, which are close to those found in polypeptide or protein (Figure 4). Just so, we referred to it as a protein-like molecule. Since the physical properties of such hydrogen bonded amide-I systems are very sensitive to bond lengths, hence, many men thought that studies of acetanilide can reveal some new phenomena. Then we here used the experimental results for the acetanilide to verify the correctness of the theory of bio-energy mentioned above.
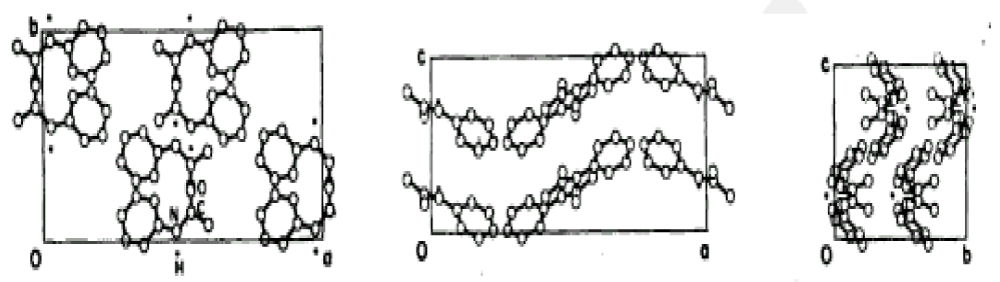
Figure 3: Various views of the cell unit of acetanilide, with cell parameters a = 19.640 °A, b = 9.483 °A, and c = 7.
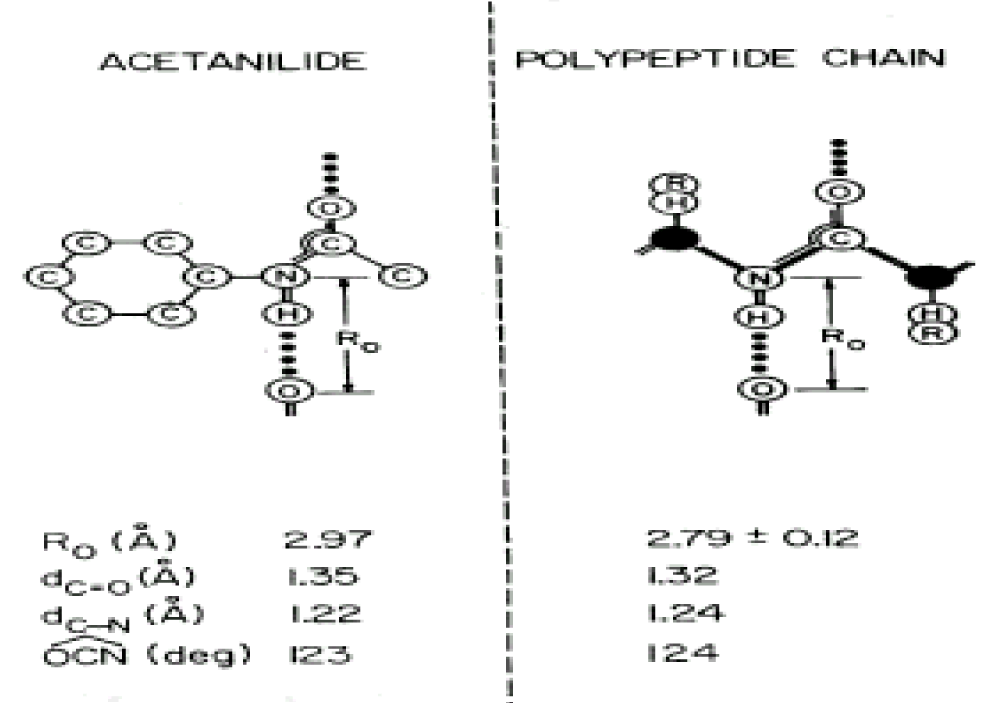
Figure 4: Comparisons between of acetanilide and a protein molecule [138-142]
The properties of Infrared absorption and Raman spectra of acetanilide
As it is known, the features of structure and conformation of the acetanilide and their changed can be easily observed and inspected by infrared and Raman spectra. Thus Careri and co-workers [137-142] collected the infrared spectra of absorption of ACN using three different infrared spectro-photometers. For the study of temperature dependence of the amide-I region, a Nicolet model 7000 Fourier-transform infrared spectrophotometer was used. Spectra were collected for 100 scans using a band width of 0.5 cm−1. The sample was thermostated using a closed-cycle helium refrigerator. Far-infrared absorption spectra were measured using a Michelson interferometer (model 720) equipped with a Golay cell. Samples consist of pellets obtained from a mixture of grounded ACN and polyethylene power. Pure polyethylene pellets were used to measure background transmission. Raman spectra were excited by a coherent radiation model 52 argon ion laser operating at 488nm or 514.5 nm, with stabilized output power of 20-200 mW. Incident light was filtered by proper choice of interference filter and its intensity was monitored using a beam-splitter and a silicon photocell, scattered light was analyzed by a Jarrel-Ash model 25-300 Raman spectrometer and detected by an ITT model Fw-130 cooled photomultiplier using photon counting electronics. They measured the infrared spectra of amide-I (1600- 1700 cm−1), amide-II (1500-1600 cm−1), amide-III (1300-1500 cm−1), amide-IV or VI (500 -700 cm−1) and amide-V (700-800 cm−1). The infrared spectra of amide-I in 1600-1700 cm−1 and 2500-3500 cm−1, 4750-4850 cm−1 and 6200-6400 cm−1 are shown in figure 5. The absorption intensity vs. temperature is shown in figure 6. The Raman spectra of amide-I in 1630- 1700 cm−1 and low-frequency modes at 300K and 50 K are shown in figure 7 and figure 8, respectively.
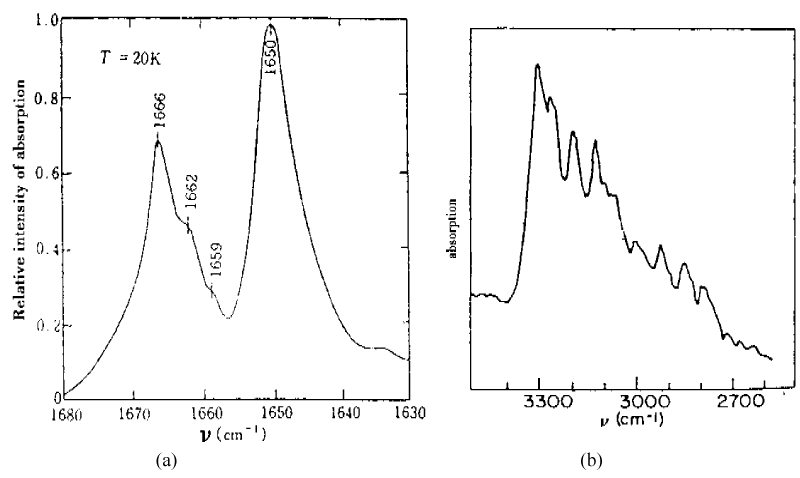
Figure 5: Infrared absorption spectra of ACN in (a) 1600-1700 cm−1 and (b) 2500-3500 cm−1 [138-142].
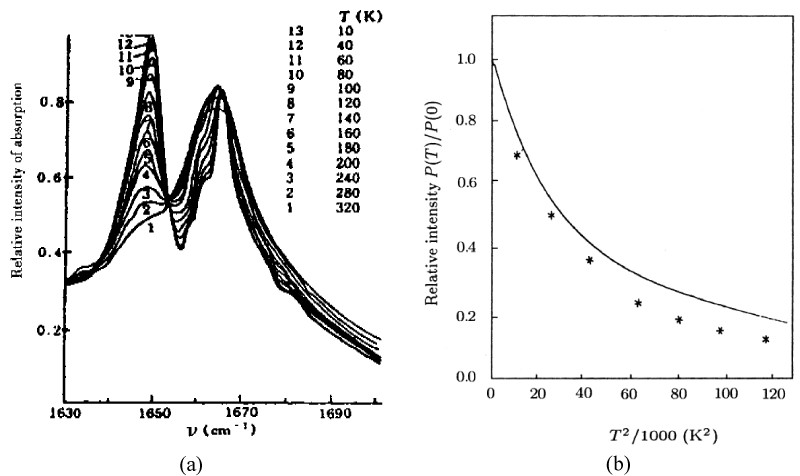
Figure 6: (a) The absorption intensity vs. temperature for ACN, and (b) comparison of experimental and theoretical values [138-142].
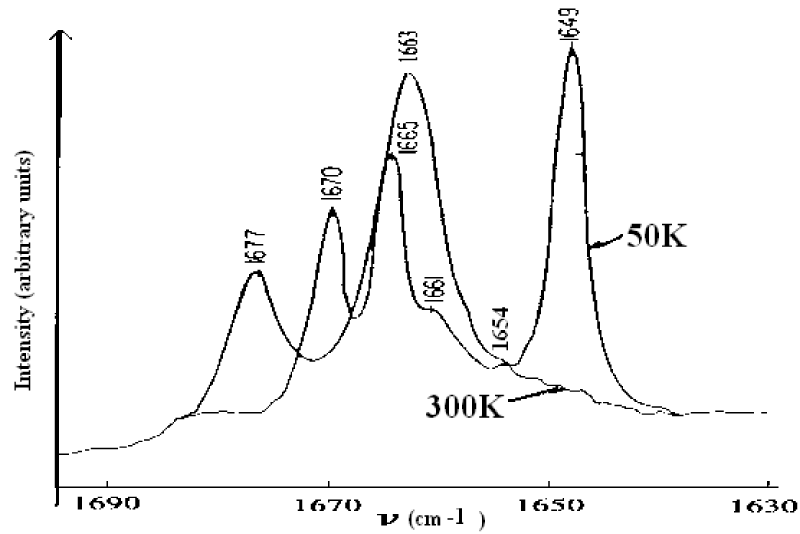
Figure 7: Raman spectra of ACN in 1630-1700 cm−1 at 300 K and 50 K, respectively [138-142].
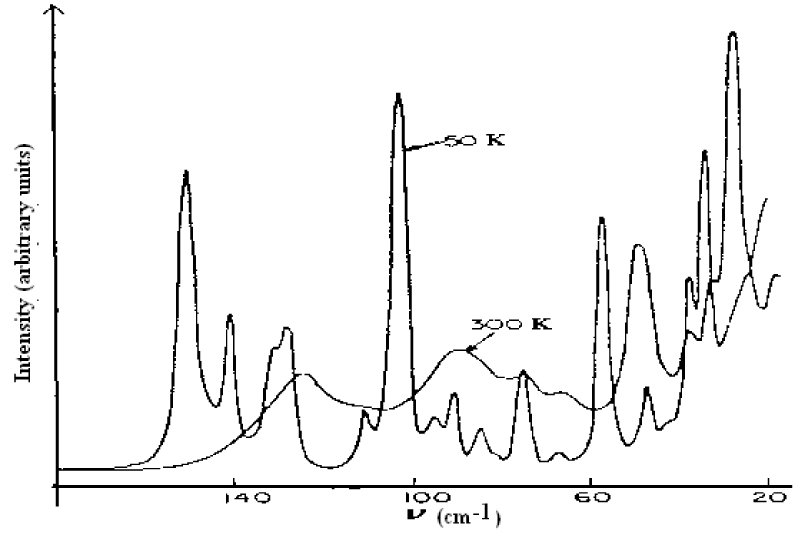
Figure 8: Low frequency Raman spectra of ACN at 300 K and 50 K, respectively [138-142].
Careri and co-workers found an anomalous line,1650cm−1, visible at low temperatures and 16 cm−1 below the amide I excitation,1666cm−1 in ACN crystal. How have we explain the results of infrared and Raman spectra in figures 5-8? This is worth to study deeply. As it is known, the characteristic feature of amide-I group, CONH, in polypeptides is amide-I mode, which mainly involves the stretching of C=O bond. This mode is observed as an infrared absorption peak at 1666 cm−1 in acetanilide and near this value in a wide variety of materials, including the amide-I groups. The corresponding spectroscopic evidence of the new band at 1650 cm−1 has been mentioned earlier, but detailed measurements of the crystal structure and specific heat as a function of temperature of peptide groups preclude assignment of the new band to (1) a conventional amide-I mode, (2) crystal defect states, (3) Fermi resonance or (4) frozen kinetics between two different subsystems. The correct assignment is the self-trapping of the amide-I vibrational energy. This conclusion is based on the following experimental facts: (1) the 15N substitution induces a small shift to the amide-I at 1666 cm−1, and the new band is also shifted by the same amount; deuterium substitution at the NH position strongly affects both the amide-I and the new band in a complicated way; (3) upon cooling a decrease in the integrated absorption of the normal amide-I and a corresponding increase in the integrated absorption of the 1650 cm−1 band are observed; (4) the 1650 cm−1 band and the amide-I band show the same dichroism over the temperature range integrated; and (5) the measurements of specific heat, the dielectric constant and the volume expansion as a function of temperature rule out the occurrence of rotational isomerism or polymorphic transition which would affect some other infrared and Raman absorption bands, but not the new band. The self-trapping mechanism of amide-I vibrational energy used by Scott and Eilbeck et al. [18,141-146], comes from the Davydov model of vibration energy transport in alpha-helix protein molecules [5-11,20,147]. They have given a good account of the properties of first excited state in this model. Thus we should assign the eigenenergy spectra of amide-I are 1665 cm−1, 1662 cm−1 and 1659 cm−1to the B2u, B1u, B3u modes, respectively, the 1650 cm−1 mode in figure 5(a) and figure 7 should be assigned to the exciton-solution in ACN, the 16 cm−1 below the amide I excitation in that crystal is red-shifted from the main peak at 1666 cm−1, which was interpreted as a self-trapped, solution state. Thus, the solution excitation indeed exists in these systems. Also, the peak values in figures 5-7 including the overtone spectrum of the vibrational solution in the infrared absorption in crystalline acetanilide obtained by Scott el al. [144], agree with the nonlinear eigenenergy spectra of the exciton-solution, which were given obtained from finding the eigenvalues of Hamiltonian operator in Pang’s theoretical model [148-151].
In Pang’s calculation the following Hamiltonian of the acetanilide [148-151] is used:
This Hamiltonian includes vibrational excitation of amide-I caused by localized fluctuation and vibration of the peptide groups. This is, in turn, caused by structural deformation of molecular chains and the interaction between the two modes of motion in the respective crystals. Here m is the mass of amide-I vibrational quantum (or exciton),
are diagonal and off-diagonal elements of the dynamic matrix of vibrational quantum,
is also a Einstein vibrational frequency of the exciton, and
is the interaction between the nearest neighboring excitons caused by the dipole-dipole interaction in molecular chains. Likewise rn and pn = m(drn / dt) are the normal coordinates of the n th excitons and its canonical conjugate momentum, respectively, M the mass of a peptide group in the unit cell,
are the change of energy of exciton and of coupling interaction between the excitons for an unit extension of molecular chain, respectively. Rn and Pn = M Rn are the canonically conjugate operators of displacement and the momentum of peptide group and β is the elastic constant of molecular chains. Hex in H is the Hamiltonian of harmonic vibration of amide-I including the off-diagonal factor, Hph is the Hamiltonian of harmonic vibration of the peptide group and Hint is the interaction Hamiltonian between the two modes of motion.
The wave function of the exciton and phonon in the system is denoted by
Where
and
are the vibron or excitonic and phononic ground-states, respectively. From Equations (19)-(20) we can obtain the dynamic equation of the exciton [78,80,117], which is
Where G is nonlinear interaction constant,
for the new model h = 1, s=v/v0, v is velocity of exciton, v0 is the sound velocity in peptide lattice. For the acetalinide containing two chains, the Hamiltonian operator of the system in second quantum representation corresponding Equation (21) is
From
we can eventually the Eigen energy of the Hamiltonian operator, which is shown in table 2. These results in table 2 denoted that the solution theory is demonstrated by these experiments.
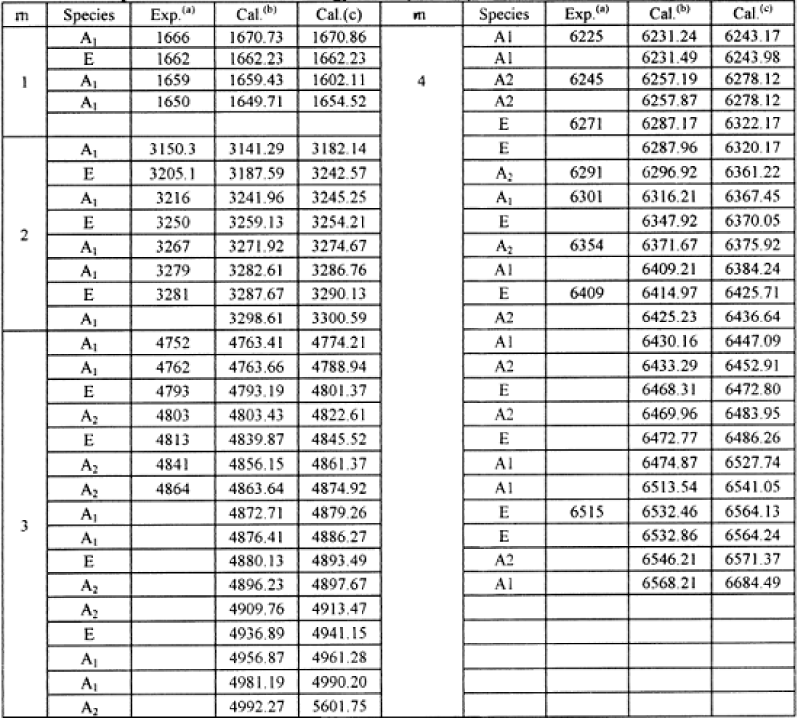
Table 2: The quantum vibrational energy levels (in cm−1) of acetanilide, where the experimental result from compilation of [138-147]. (b) the results for Pang’s model, (c), the results for the Davydov model.
In the meanwhile, in experiments of infrared absorption and Raman scattering when the crystalline acetanilide is cooled from 320K to 10 K, but no other major changes occur from 4000 to 800 , only the intensity of this new band increases steadily from room temperature to 10 K as shown in figure 6a, but it is not present in amorphous materials or acetanilide methylated at the position where hydrogen-bonded distances occur, but it is recovered after annealing.
Similar phenomena can be observed in Raman scattering experiments [5-11,138-147]. From figure 6 we know that the absorption intensity decreases in the form of
with increasing temperature, which can be obtained using the solution theory by Alexander and Krumbansl [152,153] and Scott et al. [154], as well as Pang et al. [93,95,99]. Alexander et al’s and Pang’s results are represented by
and , respectively. In Figure 6b we denoted the comparison between experimental (denoted by “*”) and the theoretical values (solid curve). This manifests that the experiment supported really the existence of the solution in the system.
The results obtained from Pang’s solution theory [78,80,117] with the above experimental data in infrared absorptions of CAN [138-147] are together shown in table 2. From this table we see clearly that the theoretical results are well consistent with the experimental data. This indicates clearly that Pang’s solution theory is appropriate to the acetanilide.
Utilizing the theoretical data in table 1 at m=2 we got the Raman lines in the acetanilide, which are shown in figure 9. The nine lines occurring in this figure, 157 cm-1, 145 cm-1, 130 cm-1, 100 cm-1, 95 cm-1, 84 cm-1, 72 cm-1, 57 cm-1, 39 cm-1 are quite consistent with the experimental data of nine peaks in the Raman spectrum of acetanilide at T=50K in figure 8 by G Careri et al. [138-147] as 154 cm-1, 148 cm-1, 127 cm-1, 104 cm-1, 92 cm-1, 84 cm-1, 75 cm-1, 60 cm-1, 38 cm-1, respectively. This shows also that Pang’s solution theory is correct.
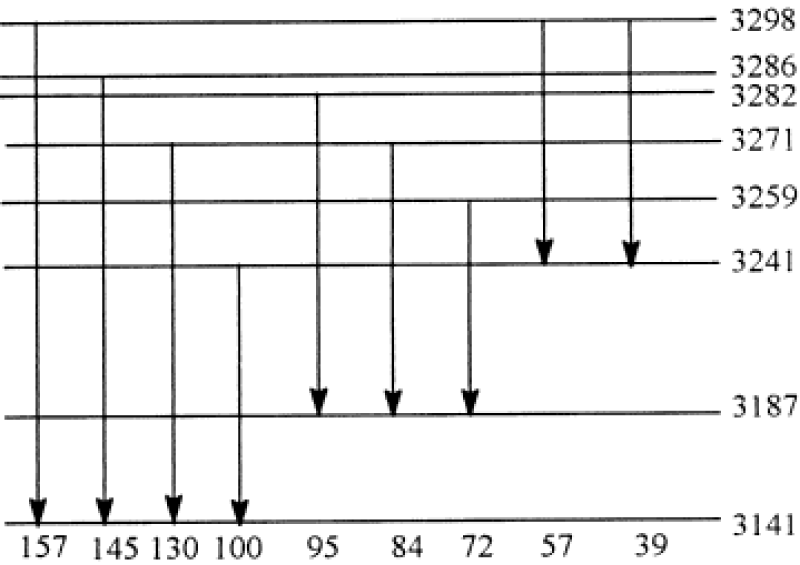
Figure 9: Distribution of energy levels at m=2 and corresponding Raman lines of acetanilide.
The properties of Infrared spectra of absorption of the acetanilide by pump- probe spectroscopy
Hamm et al. [158-164], measured the properties of infrared spectra of absorption in acetanilide by pump-probe spectroscopy. In the pump-probe experiment the various states of the system are first excited with an intense short IR laser pump pulse, and the response upon that excitation is then probed by a second short, but much weaker probe pulse. Most of the spectroscopy performed so far on the ACN problem was stationary IR absorption spectroscopy and its temperature dependence as shown in figure 10. The information gained from the pump-probe experiments, as compared to stationary IR absorption spectroscopy, is manifold. At the same time, it can measures the anharmonicities of the individual states, in which its first excited state is populated when pumping a state, and the probe pulse is probing both the upward transition to the second excited state and the downward transition back to the ground state through stimulated emission. If the state is harmonic, both upward and downward transitions appear at the same frequency, but with opposite signs, and cancel each other (if taking into account the effect of bleaching the ground state). On the contrary, when the state is anharmonic, they lead to two bands with opposite signs separated by the anharmonicity of the transition. Therefore, the pump-probe spectroscopy is particularly sensitive to the anharmonic (nonlinear) part of the molecular potentials, which is impossible for the stationary IR absorption spectroscopy. If two states are coupled, excitation of one state will result in a response, such as a frequency shift, of the other. This means that there are the entangled transitions between them. If pumping two or more coupled states with an ultrashort pulse, whose bandwidth covers these states simultaneously, then quantum beats can be observed. Hence, we can used it to measure the lifetimes of particles excited.
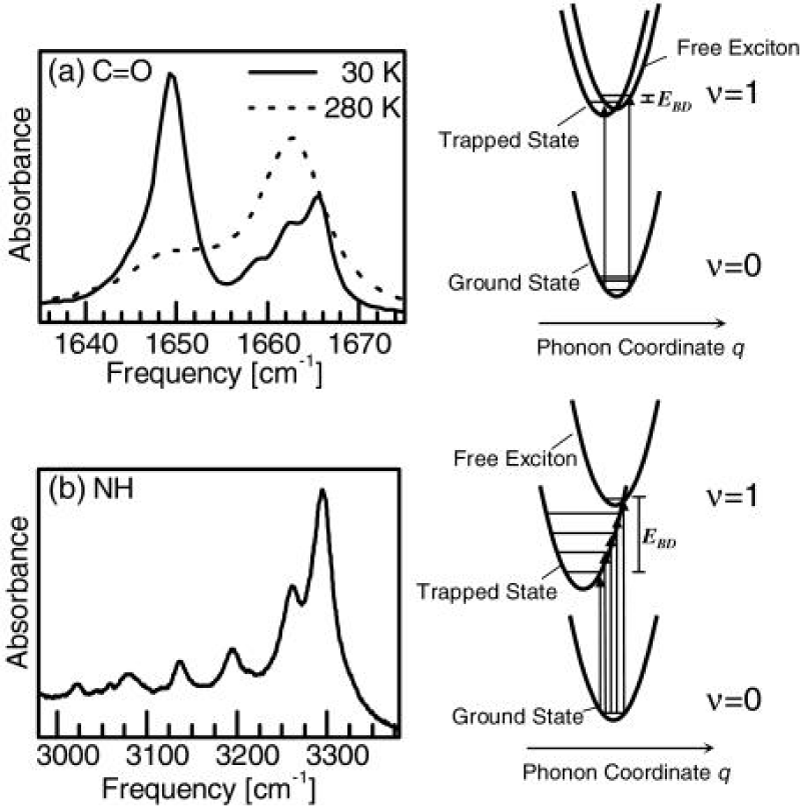
Figure 10: Infrared spectra of absorption of the C=O (a )and the N–H (b) band by pump- probe experiment in ACN and corresponding potential energy curves causing these spectra of the pumped states or soliton excited state by the femtosecond time resolution [158].
Figure 11 shows two pump-probe spectra obtained by excitation of either of the two absorption lines of ACN at 90 K, where the arrows mark the center frequency of the pump pulse. The pump pulses are spectrally narrow in this experiment (spectral width ≈14 cm−1) and excited only one band in them. When resonantly pumping the 1650 cm−1 band, it is bleached, but 1644 cm−1 band emerges. When resonantly pumping the 1666 cm−1 band, hardly any bleach of the band is observed. This indicates that the 1650 cm−1 band responds only with a signal.
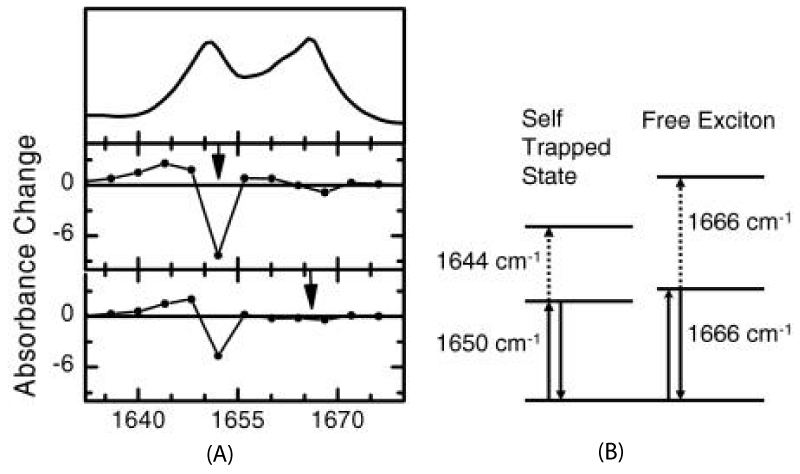
Figure 11: A is linear absorption spectrum (top) and pump probe spectra of the C=O mode of ACN at 93 K for two different narrow band pump pulses chosen to be resonant with each of the absorption bands. B is level scheme of the system, explaining the distinctively different response of both modes [161].
In the meanwhile, they discussed also the different response of both bands of ACN [159]. They started with the statement that the involved C=O vibrators are slightly anharmonic. In this case the “anomalous” band with a 0–1 frequency of 1650 cm−1 and a 1–2 frequency of 1644 cm−1 in figure 11B displays the expected anharmonic response, but the 1666 cm−1 band shows not pump-probe response at all. These results manifest that the anomalous 1650cm-1 band shows such an anharmonic response, and hence, is a localized state, but the normal 1666 cm−1 band shows no nonlinear response, which can only be the case when it is effectively a harmonic state. (it has been verified that the lifetime of the normal band is long enough to potentially observe a bleach [161]). Hence, the normal band is largely delocalized, i.e., a free exciton, in agreement with the original assignments.
On the other hand, they utilized 2D-IR spectroscopy to clarify the origin of the anomalous amide I band [162]. 2D-IR spectroscopy can distinguish between various kinds of nonlinearities of the molecular Hamiltonian. In this experiment, the pump frequency they used is continuously scanned through the spectrum, and the response is plotted as a function of both pump and probe frequencies. Figure 10 shows the comparison of the 2D-IR spectrum of crystalline ACN with those of benzoylchloride and N-methylacetamide (NMA) dissolved in methanol, respectively, where the arrows mark the position of the pump pulse. These infrared spectra of absorption display clearly the same doublet structure in all three cases, but the 2D-IR spectra differ obviously. Benzoylchloride has a prototype example of a molecule with a strong symmetric Fermi resonance [165], whereas N-methylacetamide dissolved in methanol exists in two spectroscopically distinguishable conformations, one fraction hydrogen bonded to the solvent and the other fraction not [166]. Thus their molecular structures are different from that of ACN. If the two lines in ACN come from molecules in two different conformations or surroundings (topological defects), as has been suggested various times [167], pumping of one state would not cause any response of the other state (in contrast to what is seen in the experiment). The 2D-IR spectrum should in essence be diagonal, as it is for N-methylacetamide dissolved in methanol (Figure 12). Otherwise, in the case of a Fermi resonance, original explanation of the doublet in the ACN spectrum [169], a very characteristic coupling pattern, is obtained (Figure 12). Thus, investigating these coupling patterns in detail, the compelling evidence against this possibility can be provided as well, and the self- trapping state or solution state should be substantiated [162].
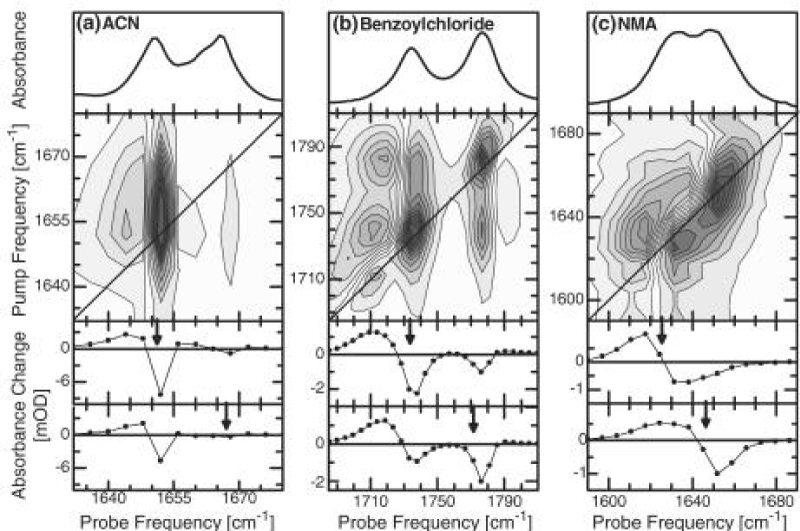
Figure 12: The linear absorption spectra and 2D-IR pump probe spectra of the C=O mode of ACN (at 93 K)(a), benzoylchloride(b), and NMA dissolved in methanol(c) (both at room temperature). Blue colors denote the negative absorption change (bleach and stimulated emission), red colors positive absorption change (excited state absorption). The two lower panels show horizontal cuts through the 2D-IR spectrum for pump frequencies resonant with either of the two bands [161].
Figure 13 shows the pump-probe response after exciting the N-H band in ACN obtained by Edler et al. [162]. In contrast to the C=O work, here they used a pump pulse so short that its bandwidth covers many of the side peaks of the N-H band. They were all excited at the same time, leading to pronounced quantum beats in the response. The Fourier transform of the signal in figure 13 gave two peaks: a dominant peak at 48 cm−1 and a weaker peak at 76 cm−1. The temperature dependence of the two peaks was obtained [162], which are completely consistent with the two lines in the conventional Raman spectrum (Figure 13) [169]. This demonstrated that the coherences indeed represent lattice phonons; in fact, this experiment identified the two phonons that mediate vibrational self-trapping in ACN. These experimental data coincide with theoretical results in Pang’s solution model [124-126].
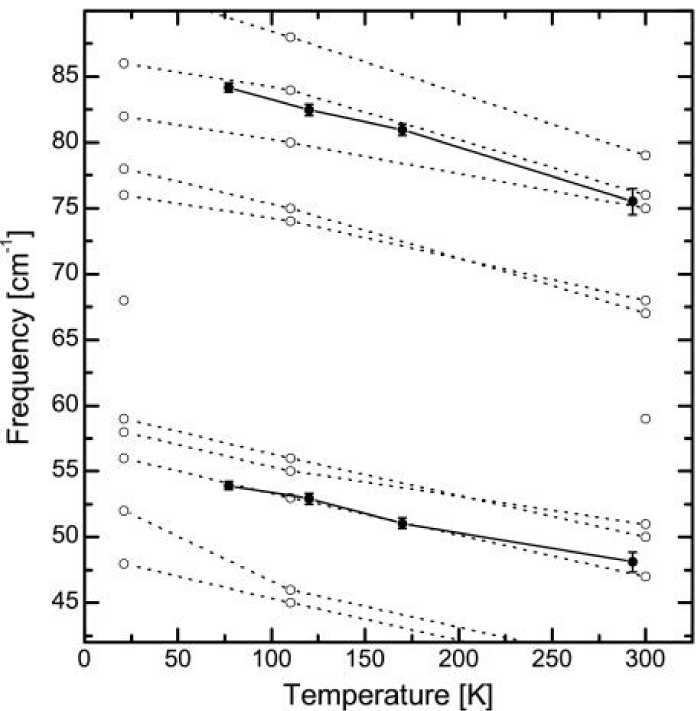
Figure 13: The temperature dependence of the low frequency Raman modes (open circles) in ACN [169] and of the beating frequencies observed in the pump-probe experiment (filled circles). The temperature dependence of the beating frequencies perfectly matches that of the Raman modes [163].
Therefore, Hamm and co-workers have identified two phonon modes that mediate the self-trapping and find further evidence for self-trapped states in ACN and N-methylacetamide (NMA) and even in a small polypeptide [164] and speculate that such states may arise in all hydrogen-bonded systems. These experimental results are very important and interesting, which provide experimental evidence to the existence of solution in ACN and confirm the validity of above theory in Pang;s model. Thus, the solution excitation, predicted by the above theory indeed exists in the acetanilide crystal.
Infrared and Raman spectra of collagen and bivine serum albumin
Infrared spectra of collagen protein: As known from molecular biology, the collagen is a most common form of protein, and a main component in musculature in surface cells, for example, smooth muscle[. In normal physiological condition it is a soft condensed matter, and resembles closely a solid state up to 95oC. The tropocollagen, a kind of collagen, is a fiber protein which has a quaternary molecular- structure with superhelix made of three channel α-helix chains and containing a sugary side chain. Each α- chain involves 1050 amino acid residues in which the glycine is about 35%, proline 10%, hydro-proline 9% as well as some alumine and hydrolysin. In order to understand clearly its molecular structure we must analyze in detail its primary, secondary, tertiary and quaternary molecular structures. Its one-dimensional (or primary) structure is a long chain made up of [Gly-X-Y]n where X and Y are proline and hydro-proline or hydrolysin, respectively. The secondary structure is a α-helical structure with left-spin, which is formed by means of regular folding along the above one-dimensional main-chains. In such a structure the glycine is located at the center of helix, the proline and hydro-proline are located at the exterior of helix. If this secondary structure is further extended along the axial direction in the manner of left-hand helix, and is again folded as several helix-structures, then the tertiary structure of collagen is formed, in which the main chain is made up of several helices. If the above three long-chains collect again together and wind each other in the manner of right-hand helix via linking by hydrogen bonds between the amide bond C=O and sub-amino group NH, i.e., C=O…NH, to form a fiber, this is just the so-called quaternary structure of collagen. In this structure the side-groups of two lynsin residues contained in each peptide chain are linked by covalent bonds under action of oxido-reductase of lynsin. Such a long chain can be represented by -CO-CH(NH)- CH2-CH2-CH2- CH = N-CH2- CH2- CH2-CH2- CH(NH)- CO-. Hence, the collagen is a kind of protein molecule with quaternary structure containing α-helical constitutions, in which the alternate arrangement of hydrogen bonds and covalent bonds play important role in stabilizing its structure, enhancing its tension strength and completing its biological-functions, such as the energy and information transports [170].
We measured the infrared spectra of collagen in terms of different methods and instruments, in which the type I-collagen is used in our experiment [106,171,172], it is extracted from the skin in mouse and further purified, in which water is drained completely. Subsequently, the samples of the collagens without water are sandwiched between KBr windows and transferred to a temperature cell in the infrared spectrometer. Their spectra of infrared absorption are recorded by a Perkin Elmer spectrum GXFFIR spectrometer equipped with a DTGS detector and having a resolution of 2cm-1 made in Germany. The light source used here is infrared silicon-carbon bars. The measurements of infrared absorption for the samples are performed in the range of 400-4000cm-1. To obtain an acceptable signal-to-noise ratio 16 scans are accumulated. The infrared spectra of absorption of collagens at different temperatures are recorded, when the sample temperatures are varied from 15℃ to 95℃ in the intervals of 10℃ with a reported accuracy of ±1℃, which is controlled by the equipments with variable-temperature, in which the water or water vapor is completely drained. The spectra of infrared absorption of the collagen from 400 to 4000cm-1 at 25℃ is shown in figure 14. The spectra at high frequencies are dominated by the vibrational modes of amide, in which a new band at 1650.0cm-1 is also found, except for there are two vibrational modes of amide-I, 1666.1cm-1 and 1680.4cm-1. At the same time, other amide bands, such as, the amide-II band at 1542.1 cm-1, amide-III band at 1455.9cm-1 and 1404.5cm-1, amide-IV band at 1335.8cm-1 and 1243. 49cm-1 and amide-V band at 1081cm-1 in the range of 1000-1500cm-1, are also found. Meanwhile, there are rich spectrum-lines in the range of 2800-4000cm-1, such as, 3433.2, 3408.0, 3389.7, 3355.6, 3296.3, 3244.1 and 3209.0cm-1, etc., which are not observed as yet. In figure 15 we give the spectrum of infrared absorption of collagen at the lower temperature, -1000C, by using this spectrometer. We see from this figure that many peaks of infrared absorption in the collagen occur in such a case.
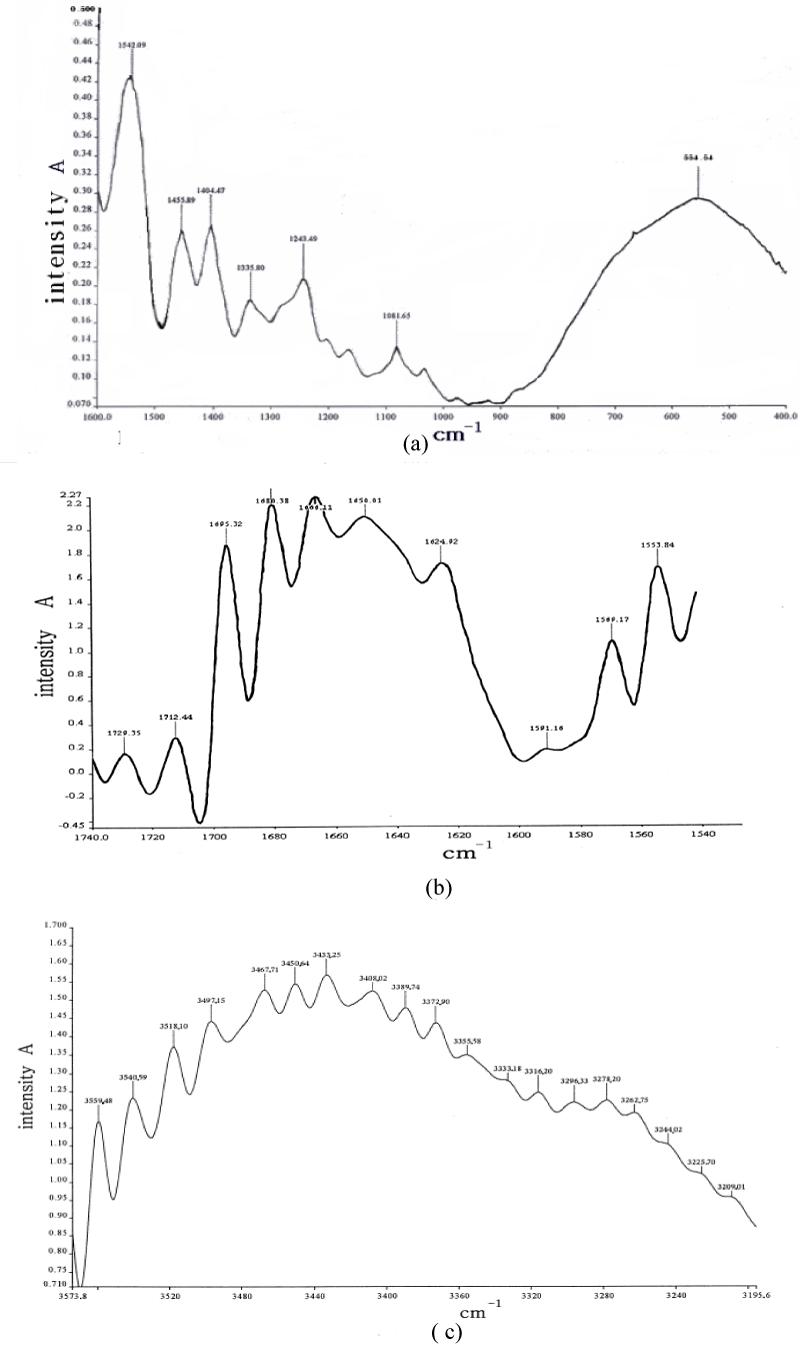
Figure 14: Infrared spectrum of the collagen from 400 to 4000 cm-1 at 25 0C.
We [106,172] are more interested in the behaviors of the two bands of 1666 cm-1 and 1650cm-1 because they related to the excitations of the exciton and solution, respectively. Thus we here study in detail their features. A detailed result of temperature-dependence of absorption intensity in the range of 15-950C is collected and shown in figure 16. We can see from this figure that the intensity of 1650 cm-1 band increases with decreasing temperature without apparent change in frequency and shape, but is only weaken at 950C. On the other hand, the amide-I infrared absorption at 1666cm-1 band decreases on cooling. The peak intensities of 1650 cm-1 and 1666 cm-1 band as a function of temperature are shown in figure 17, respectively. Clearly, different temperature-dependences of intensities for 1650 cm-1 and 1666 cm-1 can be seen. It is surprising that the absorption intensity of 1666 cm-1 increases linearly with increasing temperature, but the intensity of 1650cm-1 decreases exponentially with increasing temperature, which can be approximately expressed by
, where T is represented by Celsius temperature, 0C is its unit, I0 is a constant related to the initial intensity. Figure 17b gives the linear temperature -dependence of the relative intensity, I/I0, for the 1666cm-1 peak. Figure 17a gives the relation of logarithm of relative intensity, Ln (I/I0), vs. (T/0C)2 for the 1650cm-1 peak. From this figure we see clearly that the change of experimental data satisfies basically the linear relation of
versus the temperature T, which is similar with the rule obtained from figure 6a in CAN obtained by G. Careri et al. [138-141], and also with the theoretical results of exponential decrease of intensity of 1650cm-1 band with temperature obtained by Alexander et al. [152,153], and Scott [154] in ACN and Pang [106] in protein molecules. Therefore, from this investigation Pang obtained that true existence of the solution, which is formed by self-trapping of the excitons through its interaction with a band of optical-phonon modes clustered near 70cm-1. This means that Pang’s model gave really the experimental results of red shift of main peak and anomalous band of 1650cm-1 in the infrared absorption of the collagen. The concordance between the theoretical and experimental results support the real existence of the solution in the collagen and correctness of Pang;s model.
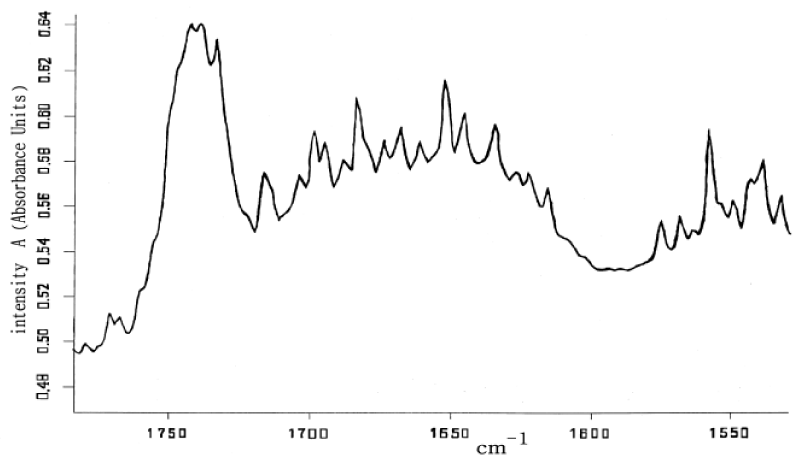
Figure 15: Infrared absorption spectra of collagen in the region of the amide-I mode at lower temperature of –100oC.
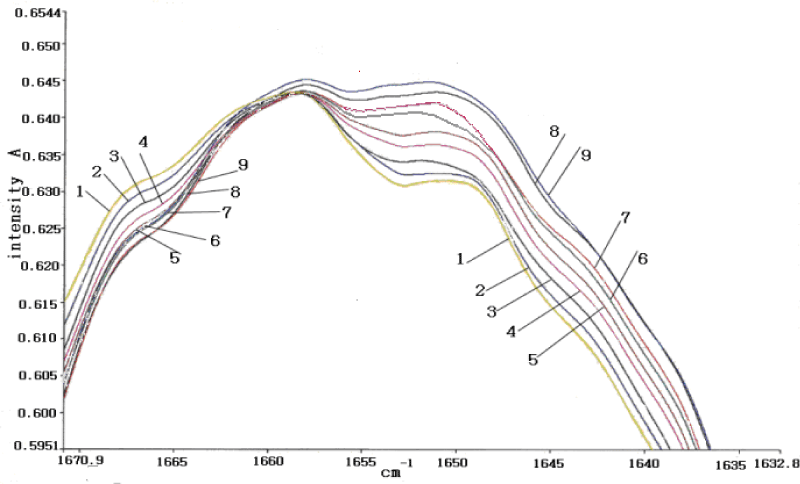
Figure 16: The infrared absorption intensities of amide-I in the collagen at different temperatures, 950C (1),850C (2), 750C (3),650C (4), 550C (5), 450C (6), 350C (7), 250C (8) and 150C (9).
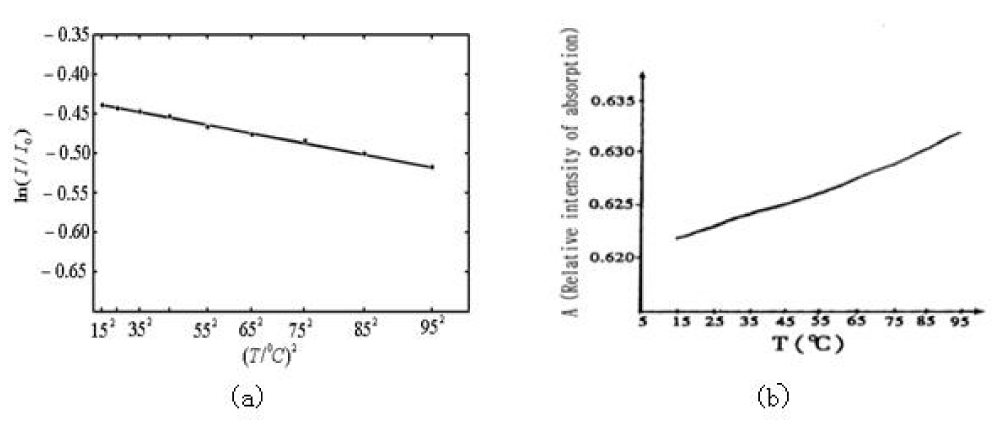
Figure 17: The temperature-dependences of intensity of infrared absorption of collagen in the region of 15 – 950C. (a) is the relation of logarithm of relative intensity, Ln(I/I0), versus (T/0C)2 and corresponding experimental data denoted by “•” for the 1650cm-1 peak, where the solid line expresses Ln(I/I0)=(0.437+8.987×10-6(T/0C)2). (b) represents the linear temperature-dependence of the relative intensity I/I0 for the 1666cm-1 peak.
In this experiment, we completely drained the water or water vapor from the tested samples and Variable-temperature bath in the FT-IR spectrometer. These showed that the above results were not related with accuracy of the instruments used and water in samples. Therefore, it is believed that the above results are credible and reliable.
Figure18 showed the laser-Raman spectra of acidity I-type fiber- collagen , which was obtained by Chu Guo-pin et al. [172], using the SpeX1403-type laser- Raman instrument made by American. We see from this figure that there are clearly 1650.0cm-1, 1666.1cm-1, 1670cm-1 and 1680.4cm-1 in collagen, which resemble those in ACN. Obviously, the peak at 1650.0cm-1 corresponds the solution, other peaks denote the vibrations of Amide-I. Thus this experiment supported again that the solution exists really in the collagen.
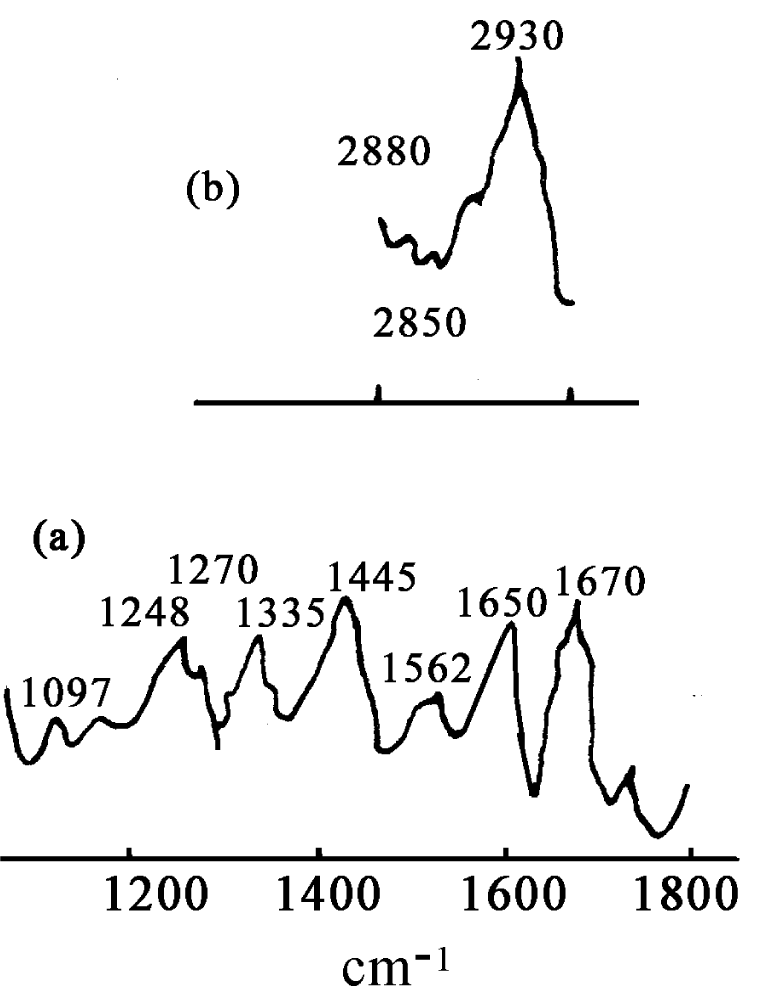
Figure 18: Raman spectrum of collagen at room temperature [17].
The peak values in figures 14-15 in the infrared absorption in the collagen can be also obtained by Pang’s theory of bio-energy transport in alpha-helix protein molecules with three channels [119,124-126]. Actually, from Equations (6)-(7) we can get
in the α-helix protein, where
=Col. (
,
,
,
,
,
,
,
,
) is a complex 9-vectors, D(|
|2) denotes the diagonal matrix, diag. (|
1|2, |
2|2, |
3|2, |
4|2, |
5|2, |
6|2, |
7|2, |
8|2, |
9|2), and
is a real symmetric matrix with nine orders, which represents various neighboring interactions between amide-Is, i.e.,
Using the method of second quantization, the complex mode amplitudes are replaced by harmonic oscillator creation and annihilation operators
and
with the properties:
, and
.
Then the effective Hamiltonian operator of the system corresponding Eq.(23) becomes now as
where the number operator is
. We assume that both frequencies and energies are measured in the same unit (cm-1). Thus we can calculate the energy-spectra of the above Hamiltonian operator in the α -helix proteins by using Equation (25) in second quantization representation, where v=0 or s=0. In this case, the wavevector of the system is chosen as: |m,k>=|m1>|m2>|m3>|m4>|m5>|m6>|m7>|m8>|m9>, where m (=m1+m2+m3+m4+ m5+m6+m7 +m8+m9) is the total quantum number, k=1,2,…, d(m), and d(m) is equal to the number of ways that m particles are distributed in the nine states. The common eigenfunction of the Hamiltonian and particle number operators is now taken as |ψm>=C1|m,1>+C2|m,2>+…+Cd(m)|m, d(m)>. Using the eigenfunction and eigenequation of the above Hamiltonian operator,
we found the nonlinear eigenenergy spectra of the solution, which are shown in table 3. At the same time, the experimental results obtained from figures 14,15 are also listed in table 3. Through the comparisons between them we know that the experimental data agree basically with the theoretical results. This indicates that the Pang theory of bio-energy transport is correct, the solution exists in the collagen with α-helical structure.
| Table 3: The vibrational energy-spectra of protein molecules with three channels in cm-1, where exp(a) are experimental values; (2) cal(b) are calculated valules in Davydov theory; (3) cal(c) are calculated values in Pang’s theory. |
| M |
exp(a) |
cal(b) |
cal(c) |
M |
exp(a) |
cal(b) |
cal(c) |
| 1 |
|
1611.01 |
1610.42 |
1 |
|
1612.95 |
1612.01 |
| 1 |
|
1628.35 |
1627.64 |
1 |
|
1631.61 |
1630.11 |
| 1 |
1650 |
1654.37 |
1653.81 |
1 |
1662 |
1662.95 |
1661.98 |
| 1 |
1666 |
1668.23 |
1667.65 |
1 |
|
1679.27 |
1678.73 |
| 2 |
3150 |
3206.33 |
3179.40 |
2 |
|
3212.17 |
3203.19 |
| 2 |
3205 |
3213.6 |
3204.71 |
2 |
|
3224.25 |
3211.85 |
| 2 |
|
3225.39 |
3212.95 |
2 |
|
3226.57 |
3213.21 |
| 2 |
3216 |
3233.34 |
3216.84 |
2 |
|
3234.71 |
3218.19 |
| 2 |
|
3246.71 |
3242.48 |
2 |
|
3248.75 |
3242.45 |
| 2 |
3250 |
3252.57 |
3249.68 |
2 |
|
3259.67 |
3258.78 |
| 2 |
|
3260.85 |
3259.87 |
2 |
|
3263.57 |
3261.77 |
| 2 |
|
3264.66 |
3260.95 |
2 |
|
3265.73 |
3262.97 |
| 2 |
|
3267.91 |
3263.67 |
2 |
3267 |
3269.99 |
3267.39 |
| 2 |
|
3270.45 |
3269.43 |
2 |
|
3278.57 |
3277.71 |
| 2 |
3279 |
3279.97 |
3278.89 |
2 |
3280 |
3282.18 |
3280.21 |
| 2 |
|
3283.91 |
3282.84 |
2 |
|
3284.75 |
3283.97 |
| 2 |
|
3286.54 |
3285.44 |
2 |
|
3287.56 |
3286.49 |
| 2 |
|
3288.24 |
3287.44 |
2 |
|
3293.14 |
3290.49 |
| 2 |
|
3299.61 |
3298.96 |
2 |
|
3300.81 |
3300.09 |
| 2 |
|
3301.73 |
3301.15 |
2 |
|
3304.95 |
3302.13 |
| 2 |
|
3310.54 |
3309.47 |
2 |
|
3311.27 |
3310.21 |
| 2 |
|
3313.24 |
3312.91 |
2 |
|
3314.73 |
3313.37 |
| 2 |
|
3322.27 |
3321.54 |
2 |
|
3323.29 |
3322.49 |
| 2 |
|
3325.11 |
3323.56 |
2 |
|
3328.47 |
3327.96 |
| 2 |
|
3331.54 |
3329.16 |
2 |
|
3338.04 |
3333.91 |
| 2 |
|
3319.17 |
3345.11 |
2 |
|
3360.61 |
3358.58 |
Infrared absorption spectra of bivine serum albumin
In order to verify further the real existences of 1666cm-1 and 1650cm-1 in the α-helix protein molecules we measure and collect further the infrared spectrum of absorption of bivine serum albumin (BSA) protein by the a Nicolet Nexus 670 FT-IR spectrometer made in USA, the sample of BSA was purchased from Sigma Chemical Co. Ltd in USA. As it is known, the BSA is an important component in the blood and is composed of α β structure, where α is α-helix type with three channels, β is β sheet. Its infrared spectrum of absorption is shown in figure 19 [106,172]. From this figure we see that there are also 1671.04cm-1, 1660.34cm-1, 1650.75cm-1 and 1639.79 cm-1 in the region of 1600-1700cm-1. In accordance with the rules of assignment mentioned above, the 1671cm-1 should be the Eigen frequency of the amide-I, 1650 cm-1 corresponds to the solution in the BSA. These results are also similar with that in the collagen, thus the 1671cm-1 and 1650cm-1 bands, which correspond the excitations of the eciton and solution, respectively, are also really existent in the protein molecule containing the-helix structure.
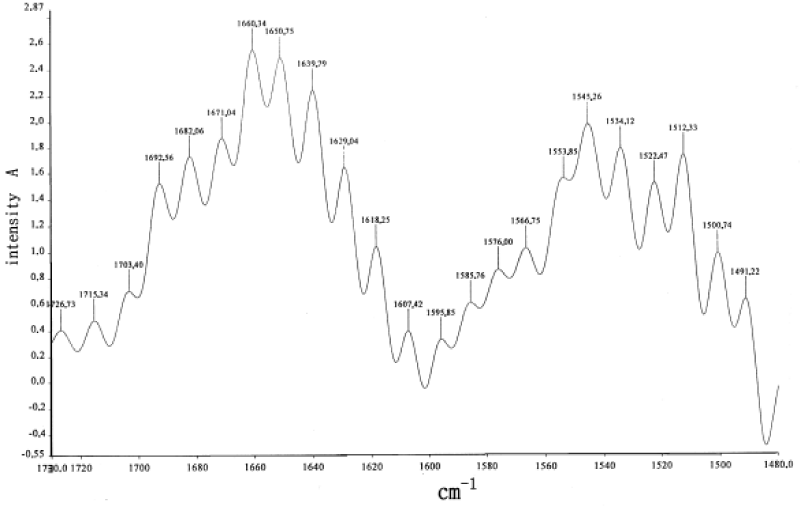
Figure 19: Infrared absorption spectra of BSA.
Infrared spectrum of absorption of myoglobin protein
It is well known that an important question in a biological context is whether the anharmonic interactions presented in the Hamiltonian of the systems exist really in proteins. Some experiments [174-177] supports strongly the true existence of the anharmonicity. In fact, the infrared (IR) spectroscopy can be used to estimate roughly the size of the anharmonicity of the α-helix amide-I bands. Upon that we inspected the temperature dependence of infrared absorbed strength. In this case we assumed that this temperature dependence comes from this self-trapping model as discussed in [18,20,138-142], in which the potential surface becomes softer with increasing energy. A truly harmonic (linear response) transition has no temperature dependence since all levels are equally spaced in energy. An anharmonic transition will show a temperature dependence with increasing temperature, then there should be a red shift in this case. Thus we can confirm the real existence of the anharmonicity through measuring the red shift of spectrum.
Figure 20 shows the temperature dependence of the amide-I vibrational spectral region of a globular protein- myoglobin (Mb) from 5 to 300K, which was obtained by Austin et al. [178-180], in which there is a clear anharmonicity in the amide-I band (and much more in the amide-II band), and a new red-shifted band appears rather similar to the acetanilide low-temperature band. If one assumes that the red-shifted band is some sort of a trapped state, as Careri and his colleagues did [138-141], then Austin et al thought that the binding energy of the state is approximately 20 cm−1, which translates into a χ of approximately 100pN, in the range needed for solution stability as discussed by Scott [142-145]. Note that the temperature dependence of the amide-I band is such that the amide-I band on the blue side of the spectrum has almost no temperature dependence. Note also that this thermal temperature dependence is different from the nonlinear optical dependence expected from driving excited states. Therefore, this experimental result is not completely consistent with that of the solution theory. We think that it could be due to the peculiarity of globular conformation of myoglobin, in which these chains are folded irregularly into a compact near-spherical shape through the hydrogen bonds, two-sulphur bonds and salt-bonds, etc., Thus the features of hydrogen bonded chains of peptide groups are different from those in general proteins with alpha-helix structure, then its features of electronic transition in this case are different from that of the fiber proteins, Upon that, its properties of infrared absorption are differs from the latter.
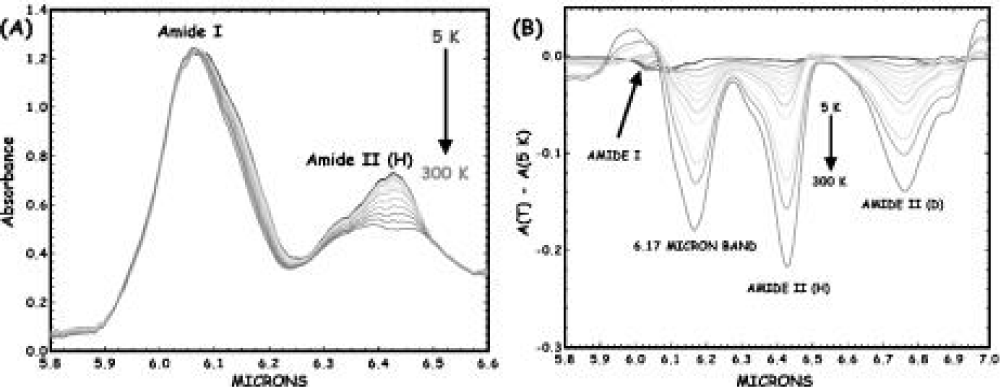
Figure 20: (A) The infrared absorption spectrum of sperm whale myoglobin as a function of temperature from 5 to 300 K from 5.8 to 6.6 μm. (B) Difference spectra of the amide I and II regions of Mb (5 K base) as a function of temperature. The band at 6.17 μm decreases with absorbance as the temperature increases [178-179]).
Raman spectrum of E. Coli.
Webb et al. [180-181], measured the laser Raman spectrum in metabolically active E.Coli. at low temperature, which is shown in figure 21. This is an interesting result. We see from this figure that there are here nine lines of spectrum of 182, 152, 123, 108, 90, 85, 63, 52 and 45-1.
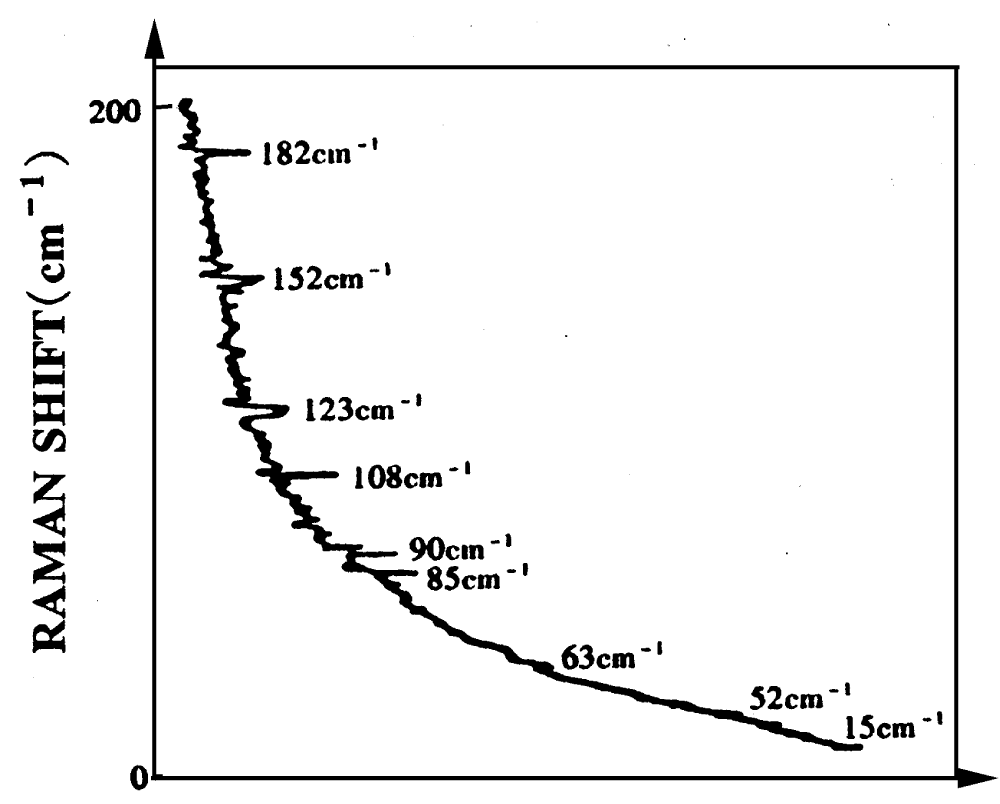
Figure 21: The Laser-Raman spectrum from metabolically active E.Coli.by SJ Webb [180-181].
Scott thought that this result provided an important experimental-evidence for the existence of the solution in the three dimensional alpha-helix proteins. Meanwhile he explained this result using Davydov theory, i.e., he thought that this is due to the result of periodic changes of bio-energy transport along three channels of the alpha-helix with a phase shift of 1200, much like the voltage on a standard three -phase power lines [142-145,182]. The frequency of alternation, v is related to the transverse dipole-dipole interaction energy, L, by
. Taking account of overtones and interactions of the moving solution with a discrete lattice, a set of internal resonances
was suggested. At about this time it became aware of some striking observations of Raman scattering from living cells c (E.Coli.) that appeared only when the cell population was metabolically active (eating, growing, dividing, etc.). These observations were a set of spectral lines, which is denoted by
. Thus he obtained
. This agreement seemed significant because
they were measured with no knowledge of the existence of the solution, and the theoretical model was constructed with the knowledge of the existence of experimental data by Scott [145,183] and later rechecked and compared with the best available experimental data by Lomhahl et al. [184]. However, Webb’s experimental results have not been verified in other laboratory [19].
We can also obtain the spectra if utilizing the energy-spectra shown in table 3, which is shown in figure 22 for m=2, where the nine lines of spectrum are 179.1, 154.5, 122.1, 107.1, 90.7, 83.9, 63.0, 55.0, 44.9cm-1, which are basically consistent with those in figure 21 [148-150]. This is absolutely not a haphazard event, it clearly manifests that the nine lines of spectrum in figure 22 are generated by transition of energy levels of quanta in the protein molecules in the chromosome of the E.Coli under active laser. Thus we also believe that the energy-spectra shown in table 3 obtained by the theory of bio-energy transport in the alpha-helix protein are relevant and correct.
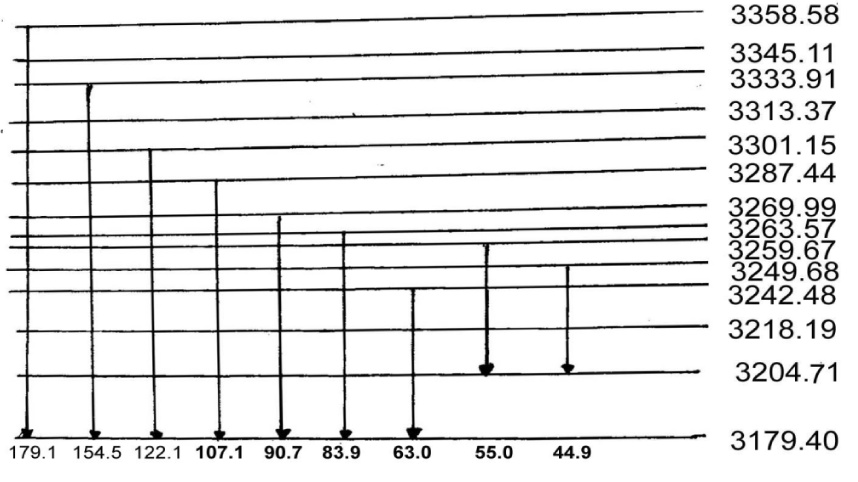
Figure 22: The distribution of energy-levels at m=2 corresponding Raman spectra of protins.
Determination of lifetime of the solution in acetanilide and protein molecules
As mentioned above, the experimental evidence for the solution excitations in proteins is strong for amide-I. However, at present, an important issue for the correctness of theory of bio-energy transport is how long the solutions last, i.e., the lifetime of the solutions should be how long to make it is a real carrier of bio-energy transport. In order to solve this issue it is very necessary to measure the lifetimes of the solution by the experiments because the lifetime of the solution are important criteria for its usefulness as a means of energy transport. Indeed, an early objection to a biological role of solution states in proteins was their short lifetimes, and thought to be in the subpicosecond range [185], but Davydov’s argument was that the solution state, having a lower energy than the delocalized, free exciton, states would last longer [5-20]. Obviously, the experimental measurement of the lifetime of solution is of interest. The experimental data so far indicate that the lifetimes of the solutions is longer, vary from system to system and is temperature- dependent, but they are comparable to the time needed for an excitation to go from a protein’s active site to another region where the energy is needed for work, according to experimental results and computer simulations. The conclusion is supported by both effective numerical simulation in [120-124] and the available experimental evidences.
The lifetime of the solution in acetanilide by free-electron laser experiment
Fann et al., measured the relaxation time of the vibrational excitation of the 1650 cm−1 band by transient-infrared-bleaching experiments which can set limits on the excitation lifetime. The source required for such measurements must be tunable around 6 μm (1650 cm−1) with adequate spectral resolution to selectively excite the band (10 cm−1). In addition, pulses with picosecond duration and sufficient intensity to bleach the transition are necessary to observe the bleaching recovery with adequate temporal resolution and signal-to-noise ratio. The source Fann et al. [186], chose to meet these criteria are the Mark free-electron laser (FEL) which produces pulses in bursts (macropulses) of about 1.3 μs in duration. The Mark III laser output is divided into pump (95%) and probe (5%) beams by a CaF2 wedge plate. Both pulses are focused by a single lens to a spot of 300-μm in diameter on the sample which is held in a closed-cycle refrigerator with CaF2 windows. The ACN was purified by repeated sublimation, crystalline domains were grown by slow cooling from an ACN melt between CaF2 disks of 2 mm thickness. A substantial increase in the probe-pulse transmission at the 1650 cm−1 band was observed when the pump pulse was simultaneous with the probe pulse. The transmission increases due to the pump roughly follows the 1650 cm−1 band shape at 0 ps delay (pump and probe temporally overlapped), indicating that the effect is associated with the 1650 cm−1 band. This show clearly that the solution excitation occurs in such a case in the acetanilide.
Figure 22 shows the bleaching recovery dynamics at the band center as assembled from many scans. The fast component in figure 23 is limited by pulse-duration, while the slow component has a decay time of 15±5 ps which can be associated with saturation recovery due to repopulation of the ground state of the 1650 cm−1 mode. The relaxation time or lifetime of 15±5 ps indicates that the 1650 cm−1 band is strongly coupled to the lattice, and the 15±5 ps is a typical relaxation value for a vibration line in the ACN. Fann et al. [186], also measured the transmission recovery of the sample at 100 ps in which the probe decays to the “cold” base line. This is an important fact since it was thought that the 1650 cm−1 band should disappear on deposition of energy into the 130 cm−1 optical phonons. Loss of the 1650 cm−1 band due to such a heating should result in an increase in transmission approximately by a factor of 2 at 100 ps, which, however, was not observed. Therefore, the time scale of such vibrational relaxations observed is inconsistent with the lifetime of 10−10 s predicted by the theory of Pang et al. [105,119,148-141,173].
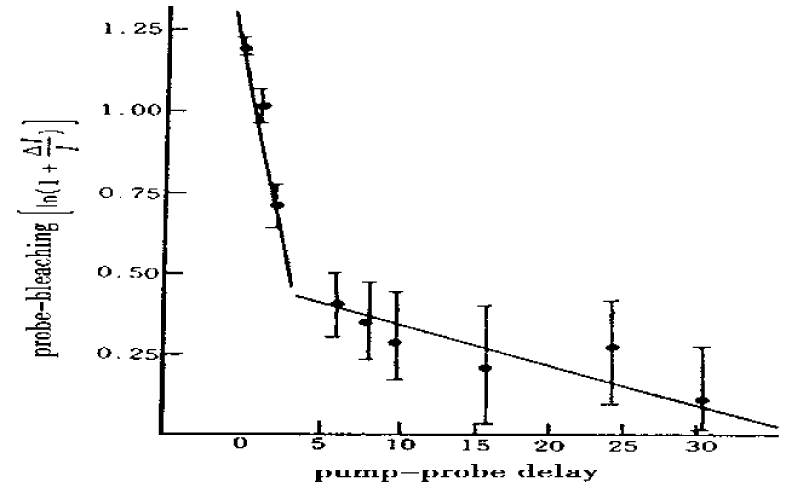
Figure 23: Probe-bleaching vs. pump-probe delay at 1650 cm −1 and 80 K ambient temperature [186].
How could this happen? Further investigation is required in order to clarify this issue. In practice, if the rate of energy flow into the 130 cm−1 optical phonon is much slower than in other materials, or the energy flows of 1650 cm−1 into the 150-200 cm−1 optical phonon, instead of 130 cm−1 phonon, the ideal time scale of 10−10 s could be observed in this experiments because the solution-optical phonon coupling can accelerate the cooling speed. Therefore, the above experiment needs to be improved.
The lifetimes of the solutions obtained by pump-probe spectroscopy
Hamm et al. [158-164], measured the lifetimes of the solutions by pump-probe spectroscopy in acetanilide and proteins. Figure 24 shows the pump-probe response of both the anomalous (1650 cm−1, filled circles) and the “normal” band (1666 cm−1, open circles) of ACN after selectively exciting the former [158]. At early times, a bleach of only the anomalous band occurs, which recovers on a fast 2-ps timescale. However, this relaxation is not complete, and a small negative signal remains. This indicates that the system does not relax back into the initial ground state, but into a state that is either spectroscopically dark or outside of spectral window. On a somewhat longer timescale (35 ps), the energy still present in the crystal thermalizes. That is, the anomalous band loses intensity (a negative signal in the difference spectroscopy of Figure 24) and the 1666 cm−1 band gains intensity, exactly as in the stationary spectra of Fig.10a when we increase the temperature. Hence, after vibrational relaxation of the initially pumped state, energy relaxes through an unknown pathway, but then reappears as heat after 35 ps. A very similar relaxation behavior was also found for the N-H band [162]. Therefore, we can determined from this experiment that the lifetime of the solution excitation in ACN is 35ps.
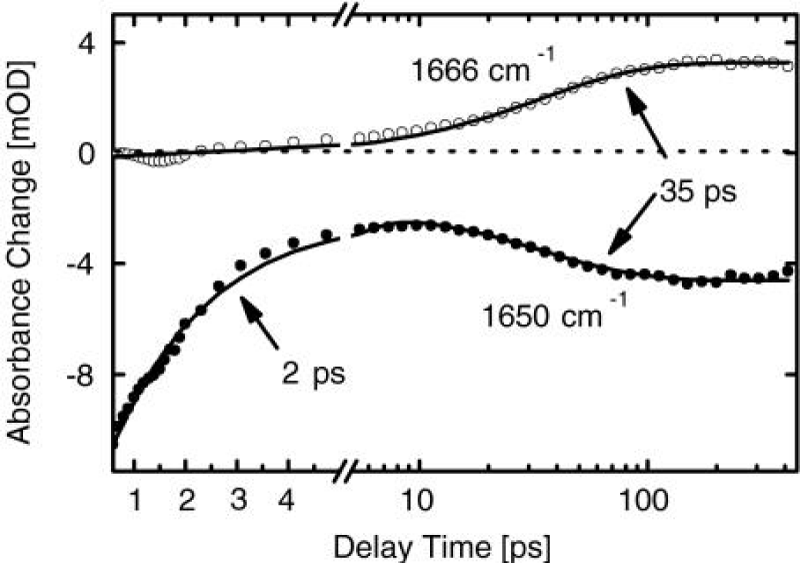
Figure 24: The pump-probe response of the anomalous (1650 cm −1, filled circles) as well as of the normal band (1666 cm −1, open circles) of ACN after selectively exciting the former [158].
Hamm et al., provided also a compelling evidence for vibrational self-trapping in NMA, which is similar with that in crystalline ACN because the former’s molecular structure resemble ACN’s. In particular, both crystals, ACN and NMA, have an orthorhombic structure and consist of quasi-1D chains of hydrogen-bonded peptide units (-CO-NH-) with structural properties that are similar to those of α-helices. Thus they are often regarded to be the model compound for peptides and proteins. However, the mechanism is expected to be generic and should occur in this crystal. Nevertheless, no convincing experimental evidence for self-trapping in NMA had been found so far. However, they carefully measured infrared spectra of NMA [161,187] by pump-probe experiment and compared the infrared absorption spectra and the pump-probe spectra of the amide-I and NH modes of acetanilide (ACN) and N-methylacetamide as well as their deuterated derivatives. The amide-I bands of NMA shows a temperature-dependent sideband, but it is less distinct than in ACN (Figure 25). On the other hand, the N-H band of NMA is accompanied by a sequence of satellite peaks, the spacings between which are larger than in ACN (which could be since NMA is lighter, and hence since lattice phonons are tentative at higher frequencies). Comparing the pump-probe responses of these spectral anomalies in both crystals gave very similar results, providing evidence that vibrational self-trapping is indeed a common effect in hydrogen-bonded crystals.
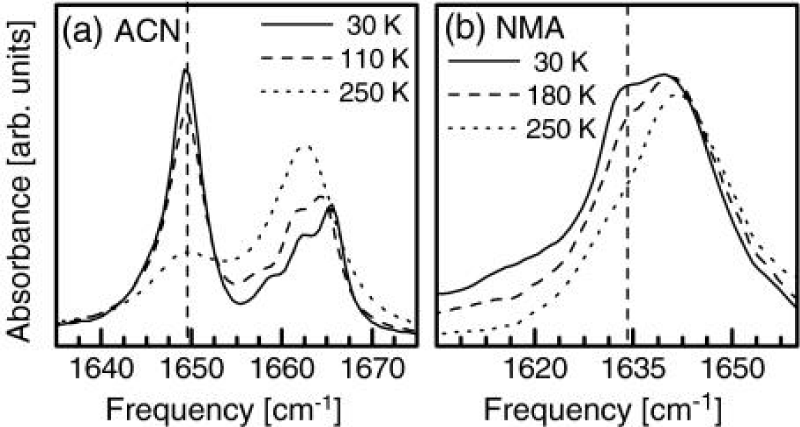
Figure 25: IR spectrum of the amide-I band in ACN (a) and NMA (b) at three different temperatures. The vertical line marks the position of the temperature-dependent sideband [161].
In the meanwhile, Hamm et al. [158-164], addressed further the question of whether vibrational self-trapping can also occur in α-helices, one of the most important secondary structure motifs in proteins. Thus, they chose poly-γ-benzyl-L-glutamate (PBLG) for this investigation because it forms extremely stable and long α-helices. The monomeric unit of PBLG is a nonnatural amino acid with a long side chain that stabilizes the helix; however, the helix backbone is identical to that of natural α-helices. Because of its stability, PBLG has served as a standard model helix [162].
They performed also an experiment which comes very close to one originally proposed by Knox [188]. They measured the heat conductivity of a peptide helix by depositing locally large amounts of vibrational energy at one end of the helix and putting certain isotope-labeled vibrational chromophore, which they employ as local thermometers, at various distances from the heater [189,190]. They found that the heat diffusivity along the helix only slightly exceed that into the surrounding solvent. However the peptide helix is not a particularly good heat conductor.
Estimation of the lifetime of Self-trapping state in myoglobin by pump- probe technique
Austin et al., estimated the lifetime of the Self-trapping state in myoglobin by pump-probe technique. In this experiment, they used 1.5 ps (10 cm−1) pulses to enhance the time resolution of the pump-probe signals [175,191,192]. They thought that if there is indeed a long-lived (say, greater than 20ps) self-trapped coherent state, then the dynamic state generated is of very narrow line width and will not be easily seen using a spectrally broad pulse. Thus, in the interest of obtaining higher spectral resolution at the cost of time resolution, they changed the pulse width to ~10ps, or ~1 cm−1 resolution.
The result narrowing the pump line width was quite shocking: on the “blue side” of the amide-I band, where there is no temperature dependence of the absorption band, there was an apparently huge (50%!) transmission change observed at 6.05μm, as opposed to the 5% transmission changes seen at the same pulse energy with a 1.5-ps pulse width at the same 6.05-μm wavelength. This huge signal is highly wavelength- dependent, scanning across the absorption band of the protein reveals, as is revealed by scanning across the absorption band of the protein. Figure 26 shows that the large signal occurs only in the spectral region where the amide-I absorbance is not temperature dependent. Therefore they thought that there is no long-lived state for the long pulse signal, but rather what looks like the convolution of the probe pulse with the pump pulse, so it would appear that what seems to be an exciting signal reminiscent of what is expected within the context of solution physics is some sort of a degenerate four-wave mixing artifact [193-196]. That is, the coherent superposition of the pump with the probe pulse on a sample whose absorbance is a function of intensity generates a diffraction grating, which scatters the pump beam coherently (diffracts) into the probe beam.
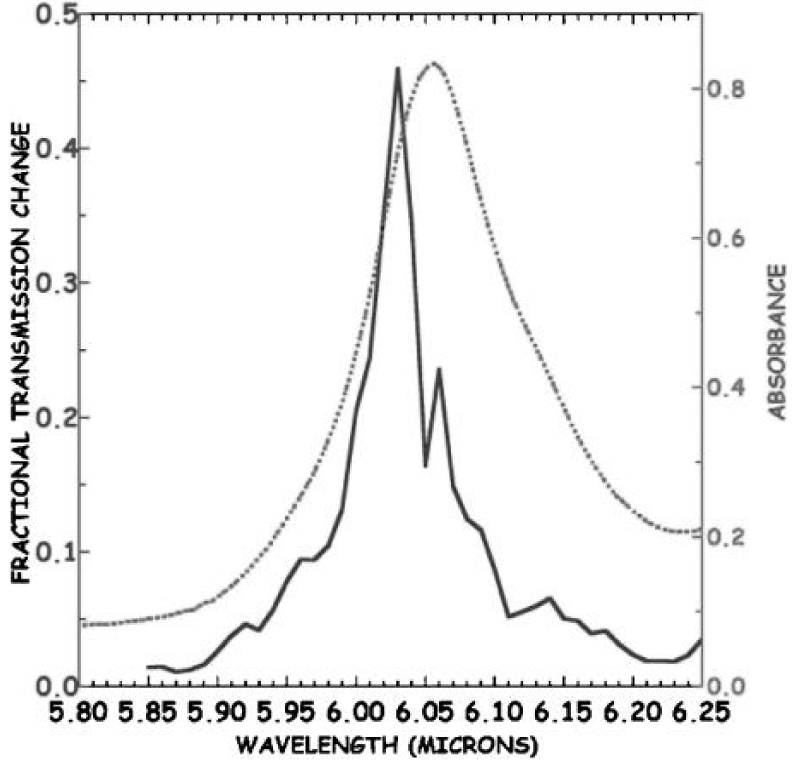
Figure 26: Pump-probe signal in the frequency domain seen with 1 cm −1 linewidth pulse scanning through the amide-I band of Mb at ∆t = 0 ps relative to the pump pulse. The sample absorbance is on the right, the left axis is the equivalent transmission change [193].
While it is consistent to assume that the large pump-probe signal is not a long lived state but rather a grating artifact on top of a short-lived saturation of the system, this grating-induced four-wave mixing explanation for the enhancement does not explain why this large signal only occurs on the temperature-independent short wavelength side of the amide-I absorbance band, and only when the pulse width is narrowed. Evidently, the grating is not due to the temperature dependence of the amide-I absorbance, since that would result in a large signal only on the long wavelength side of the amide-I band where there is a large temperature dependence to the absorbance. The grating must be due to a population depletion due to Rabi pumping of a coherent vibrational excited state. In this case there are two parameters that characterize a coherent excited state: the longitudinal relaxation time T1 which is a measure of the lifetime of the excited state, and the transverse relaxation time (pure dephasing time),
which is a measure of how long the system remains in phase [194-196]. It could be that the dephasing time of any coherently excited vibrations can be much shorter than the excited state relaxation times and determine the linewidths of the transition. In this case, it is a mistake to ignore a short
, for many nonlinear effects need the coherence times of the excitations to be long as well to be effective. A short
can be as destructive as a short T1 for a beautiful theory of nonlinear effects in biology.
The determination of T1 and
of a state starts with the static spectroscopy of the state. If a particular transition is not heterogeneously broadened, Austin et al gave the static Lorentzian linewidth Γo of the transition [193,194] by
. The classic problem in proteins is that static spectroscopy cannot disentangle T1 from, and further that the vibrational lines in the IR spectra of a protein are in homogenously broadened due to the complex energy landscape of a protein [194-196]. It is possible that within this distribution of lines there exist a set of states that are both spectrally narrow and have long dephasing times. In order to discover these states it is necessary to probe their dynamics by some form of site-selective spectroscopy.
The lifetime of the coherent state creating the grating could be significantly longer than observed if the dephasing time
of the presumably heterogenously broadened collection of states is sufficiently shorter than the true T1. To test this, they did a photon echo experiment to determine if the dephasing time was shorter than the relaxation time. The photon echo experiment was done by an OPO system. The pulse width on this system was a fixed at ~2 ps. Figure 27 shows that scanning at fixed echo delay of 0 ps with a 2 ps wide pulse across the amide-I band did indeed reveal an enhanced echo amplitude at the same wavelength where the pump-probe experiment showed large enhancement, the echo was seen only on the short-wavelength side of the amide-I band, at the same position of the enhanced pump-probe signal. However, a scan along the time axis of the photon echo, shown in figure 28, reveals that the echo has no long decay times, and that whatever state is generating this coherent signal is short-lived. Thus they finally thought that the self-trapping in globular protein of myoglobin (Mb) is not a long lived state. Very sorry, they have not given a determinate values of the lifetime from this experiment. We think that the above phenomenon is due to the peculiarity of conformation of myoglobin, which is a globular conformation, in which different bonds and groups and their interactions influence possibly the excitation and motion of the solution and excitons in the hydrogen bonded chains. Thus the self-trapping state is different from those in standard alpha-helix proteins.
Figure 27: The photon echo observed from a simple two-pulse photon echo experiment at delay =0ps as a function of wavelength. The scale of the echo pulse amplitude is arbitrary, and we have superposed the Mb IR absorbance of the amide I band [195].
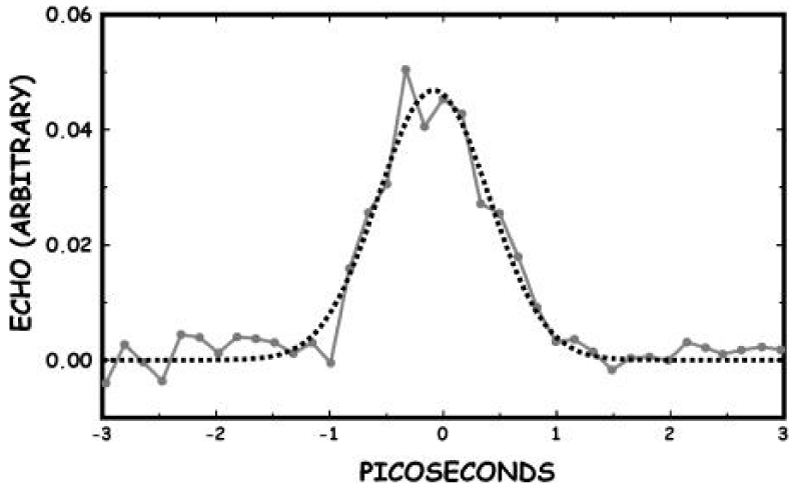
Figure 28: The amplitude of the 6.07-μm echo amplitude vs delay time. The dashed line is a fit to a Gaussian [194].
Comment for the measurement of lifetime of the solutions
Summarily, the lifetime of the solution in protein molecules and acitanilide were extensively measured and many results have been obtained, for example, Fann et al., experiment indicated that the lifetime of the solution is (15 ±5) ps in ACN [186], Austin et al. obtained [175-179], the lifetime of the solution to be 20ps in Mb, Hamm et al. [158-162], measured its lifetime by femtosecond IR pump-probe spectroscopy in acetanilide and protein, which is about 35ps. Therefore, we can say that the lifetimes of the solution are different for different systems and measured methods. In this case, how do we understand the physical meanings of the lifetime values of the solution measured? Can we whether affirm or deny the real existence of the solution and the correctness of the theory of bio-energy-transport in protein molecules utilizing these lifetime values? As a matter of fact, there are the disputations for the explanation and assessment of these lifetime values of the solutions in the bio-energy transport theory. Some persons thought that the values of lifetime mentioned above are smaller for the solution to be a real carrier of bio-energy transport in proteins because the solution cannot transport over a real protein molecules, which are composed of more than 100 amino acid residues, in the lifetimes. Thus they deny the real existence of the solution in proteins and require to measure further and again the lifetime of the solution using new method and techniques. However, others, including author, thought that the above reasons, arguments and conclusions are incorrect, we cannot deny the real existence of the solution in proteins at all by using the above values of lifetime of the solution. In the contrary, they affirmed just the exact existence of the solution and validity of Pang’s theory in protein molecules from the values. Why?
As a matter of fact, the key indexes judging the correctness of the theory of bio-energy transport in protein molecules are not the lifetime of the solution, but the numbers of amino acid residues transporting over by the solution in its lifetime. This means that the correctness of bio-energy transport depends on both the speed of solution transport and the time, in which the solution travel over one amino acid residue spacing in biological protein molecules at physiological temperature 300K because we can affirm and determine correctly the numbers of amino acid residues transporting over by the solution using only the latter, but not the former, in its lifetime. This is due to the fact that only if the solution can transport over more than100 amino acid residues, which constitute a real protein molecule, by certain speed in the lifetime, then we can confirm that this theory is correct, or else, it is incorrect. Therefore the combined data of the lifetime and velocity of solution transport and its time travelling one amino acid residue spacing, instead of the lifetime, can determine the correctness of the solution theory of bio-energy transport. This implies that the focus of disputation is not the real existence of the solution, but whether or not the solution has an enough long lifetime to perform perfectly a bio-energy transport over 100 amino acid residues. This demand us should first decide the values of velocity of solution transport and its time travelling one amino acid residue space in the biological alpha-helix proteins.
We know from Equations (1)-(15) that the solution excited in the protein molecules moves or transports in a subsound velocity v in both Davydov’s and Pang’s solution, i.e., v < v0. In which the temperature effect of medium were not considered at all. Therefore, these solutions are called subsound solution, where the sound velocity in alpha-helix protein molecules is determined by v0=r0(M/w)1/2, and the time travelling one amino acid residue spacing at the sound velocity v0 is denoted by τ0=r0/v0 = (M/w)1/2, which are v0=4.5×103m/s and τ0= 0.96×10-13 S, respectively, which are obtained from the parameter values of one amino acid residue, which are M =5.73×10-25kg= 114×3 amu (atomic mass units), elastic coefficient of protein molecular chains, W=39N/m, and the distance between the amino acids,
. Utilizing these data we can determined the number of amino acid residues, in which the solution transport in the above lifetimes in the sub sound velocity. For example, if the solutions move at three tenths of the sound velocity in the protein molecules, then it can only travel over 45, 60 and 105 amino acid residues in the lifetimes of 15ps , 20ps and 35ps, respectively. Then we determined that the lifetime of 15 ps is small for the bio-energy transport in proteins, but the lifetime of 35 ps is enough to meet the above criterion. However, if the solution moves at eight tenths of the sound velocity in the chains, then it can travel over 120,160 and 280 amino acid residues in the lifetimes of 15ps, 20ps and 35ps, respectively. Thus these lifetimes of solution are enough large for the bio-energy transport, in which the solution can travel perfectly over a real biological protein molecules. This means that the theory of bio-energy transport is correct in this case, although the temperature effect of the protein molecules has not been considered. Thus, in accordance with these results we neither affirm nor deny the real existence of the solution and validity of the theory of bio-energy transport in the protein molecules by the above results of lifetimes obtained from experiments although we can confirm the real occurrence of the solution in protein molecules from these experiments of measurement of infrared and Raman spectra mentioned above. Upon that, we, at present, have not any reasons to deny the real existence of the solutions and correctness of theory of bio-energy transport in proteins.
However, we cannot forget that these experimental results mentioned above were obtained at room temperature or physiological or finite temperatures, instead of zero temperature. At the same time, all biological protein molecules work all in physiological temperature of 300K. Then we cannot use the velocity of the sub sound solution to judge the correctness of the theory of bio-energy transport at all. This means that the influences of the temperature of protein molecules on the features of the solution must be considered in determination of its lifetime because the velocity of motion of the solution and its lifetime are very sensitive to the temperature our studies showed. In order to verify this point it is quite necessary to investigate the changes of property of the solution transporting the bio-energy are4sing from the biological temperatures, which are described as follows.
The feature of super sound movement of the solution in the protein molecules with biological temperatures
We can demonstrate that the solution transports along protein molecular chains in super sound velocity in this case due to the assistant effect of temperature on the solution in virtue of the interaction between the solution and heat phonons. In this case we must consider the thermal excitations of the phonons generated by displacement of amino acid residue arising from the temperature. Then the displacement of amino acid residue or the vibration of molecular chains must be quantized through introducing the standard transformations:
where q=2(W/M) 1/2 sin(r0q/2) is the frequency of a phonon with wave vector q, N is the number of amino acid in the molecular chain, r0 is the distance between two amino acids, are the creation and annihilation operators of the phonon, respectively.
Applying Equation (26), the Equation (7) in Pang’s model [104-126] becomes
Applying Equation (26), the Equation (7) in Pang’s model [104-126] becomes
Since the protein molecules we now study are always in biological temperature of T≠0, then there are thermal excitations of phonons in this case in the protein molecules. If considering again the quantization effect of phonon in Equation (6), then, the wave function describing the state of displacement of amino acid residues in Equation (6),
is now represented [80-81,104-126] by
Where is the phononic complete set which represents the
Elementary excitation state of single phonon due to the finite temperature T≠0. Here
(t) and
(t) are represented by
Utilizing Equations (5),(28)-(29), and the following formulae of operators in the Heisenberg representation [80-81,104-125],
We can obtain the equations of motion for the exciton and phonon. However, the protein molecules we study here are in an equilibrium state with a thermostat having a physiological temperature of T≠0, thus after finishing the calculation of the expectation value in quantum mechanics for Equation (30) we should calculate the thermal mean values of these quantities using the formulae:
The horizontal lines in Equations (30)-(31) represent just to find the thermal mean value. The density matrix,
, containing the effect of temperature, is given by
Where the diagonal matrix elements of the Hamiltonian are expressed by
Inserting Equations (27)-(28) and (31)-(33) into Equation (30), and making use of the following relationships:
We get, after some tedious calculations, the following equations of motion for the exciton and photon in the molecular chains [80-81,104-125]
From Equations (36)-(37) we obtain
Taking into account the relationship between the un (t)=<αv| Rn(t)| αn> and its Fourier variable unq(t):
Where, and inserting Equation (39) into Equation (38), we can get
In this calculation the representation:
Is used, where v0=(W/M)1/2r0 is the sound speed, and the long wavelength limit:
sin(r0q)~r0q, sin(r0k/2)~r0q/2 , sin[r0(q-k)/2]~r0(q-k)/2 ,[exp(ir0q)-1]~ir0q
Are also used. Now multiplying the two sides of Equation (40) with and summing over the
Wave number, q, at the same time, making again the continuum approximation: and utilizing further Equation (39) and
We finally obtain
The equations (41)-(42) are a complete set of equations of motion for the exciton and phonon excited in the protein molecules at physiological temperatures of T≠0. Obviously, it is a nonlinear Schrödinger equation in this case, but it’s the solutions depend closely on the temperature T of the protein molecules.
Now let =x-vt, thus the solution of Equation (41) is of the form
Inserting Equation (26) into Equation (25), we can get
Equation (44) is just the dynamic equation of exciton-solution in the protein molecules with physiological temperature, it is also a standard nonlinear Schrödinger equation, but different from Equation (11) because its parameters are all related to the temperature. When T=0, equation (44) becomes as Equation (11). Thus we can affirm that the states and features of the exciton-solution are changed relative to those in Equation (13). Then the solution solutions of Equations (43)-(44) are easily obtained, which are represented by [112-126] and
Where
The solution in Equation (45) indicates clearly that the solution generated in this case transports along the protein molecules with physiological temperature of T≠0 in supersound velocity (v>v0, s=v/v0>1), instead of subsound velocity, which is shown in Equation (13). Therefore, the states and features of the solution, which transports the bio-energy in protein peoteins with finite temperature are different from those in this case of without temperature. This means that the interaction of the excitons with the thermal phonon in the system promotes and accelerates the motion of the exciton-solution due to absorbing of the thermal energy of the protein molecules. This is just the mechanism of motion of supersound velocity of the exciton-solution formed in the protein molecules with physiological temperature. This is a new physical phenomenon. In this case this supersound solution formed is still stable because its rest energy is always lower than the bottom of the free exciton band about (1/3)Jµ2(T) [104-126], i.e., the energy gap in the energy-spectrum of excited state in the molecular chain determines the stability of the supersound solution. Also, this solution state in Equation (45) describes a quasiparticle consisting of the exciton and deformation of protein molecular chains. It thus already includes the interactions of the excitons with the vibration of amino acids (or phonons). Then, the destructin of the solution requires to remove the deformation of molecular chains. That is, in order to split the solution into an exciton and an undeformed chains it is necessary to expend sufficient energy to allow a transition from the solution state to a free exciton state. The transition probability to a molecular chain state without distortion is proportional to a Frank-Condon factor. Obviously, this is impossible in this case. Thus we can confirm that the supersound solution is stable in protein molecules with physiological temperature.
At the same time, Davydov et al. [9-11], obtained also the supersound solution solution in finite temperature in protein molecules. Therefore we can affirmed that the solutions excited in the biological protein molecules with a physiological temperature of 300K move and transport in a supersound velocity due to the fact that the interaction of the exciton-solutions with the thermal phonon and thermal energy of the proteins promote, which accelerates the motion of the exciton-solution in virtue of transforming and absorbing of the thermal energy of the protein molecules.
Since the solution transports in the suppersound velocity in this case with temperature, thus the suppersound solution can travel, at least, over 150, 200 and 350 amino acid residues in the lifetimes of both 15ps, 20ps and 35ps, respectively. This manifested clearly that the lifetimes of solutions of both 15ps , 20ps and 35ps obtained by Fann et al, Austin et al and Hamm et al are enough large for the bio-energy transport, in which the solution can truly travel over a real biological protein molecules, in which the numbers of amino acid residues are great than 100. Thus it is completely incorrect that present measurements of lifetimes of the solutions rule out the real existence of the solutions in protein molecules due to incorrect understanding and calculations [177-179]. In the contrary, we can conform completely that the solutions are really existent in protein molecules and the experiments obtained by Fann et al. [186], Austin et al. [173-179], and Hamm et al. [158-162,189-191], do really provide an enough experimental evidence for the correctness of Pang’s bio-energy transport in protein molecules.
The evidences of specific heat of acetanilide and proteins
Careri et al. [138-147], measured the specific heat of powdered crystalline ACN from liquid nitrogen to room temperature. The measured data were then fitted to the formula
In units of
, where T is temperature in ℃.
In the meanwhile, Meanwhile, Mrevlishvil [197] and Goldanskii et al [198] measured the specific heat of various biopolymers including proteins and DNA. They obtained
, which resembles Equation (48).
The temperature-dependence of specific heat can be confirmed by the solution theory of bio-energy transport in protein molecules and CAN with the teperature. In this case we have to study the properties of thermal movement of solutions and bio-enertgy transport in protein molecules and ACN under influence of temperature of medium for finding out their specific heat.
The investigations manifested that the temperature of medium will result in anharmonic vibrations of amino acid residues or peptide groups and the excitations of thermal phonons in these systems. Thus we must use the thermodynamical and statistical theories to study the influences of anharmonic vibrations of amino acid residues and interaction between the thermal-phonons and exciton-solution on dynamic properties of the exciton-solution. In accordance with this idea and thought we can find out the dynamic equation of the exciton-solution in this case.
For Pang’ solution model [104-126] in protein molecules the solution solutions are denoted by Equation (45)-(46). In this case the energy of the supersound solution or system can be also found, which is
Thus the specific heat arising from the thermal motion of the solution in the protein molecules is
At high temperature, i.e.,
from Equation (47) we have
Then we can find out the specific heat of protein molecules, which is of
Where
Where T is the absolute temperature. For the protein molecules we can obtain
using the parameter values mentioned above. Clearly, this relationship in Equation (50) is consistent with Meanwhile, Mrevlishvil and Goldanskii et al experimental data in proteins and DNA [197-200].
For the acetanilide, we can also find out its specific heat, which is similar with Equation (50) but
. Thus the experimental result given in Equation (48) in ACN is theoretically confirmed by Equation (50).
As it is well known, the correctness and availability of theory of bio-energy transport in protein molecules must be eventually verified and assessed by the experiments. Therefore, the experimental evidences play a key role in judgment of validity of this theory. In this review we collected, summarized and reviewed systematically and completely the progresses of investigation of experimental evidence for the existences and lifetimes of the solution, which is the carrier of the bio-energy transport, in past 30 years in the around world, in which we comment mainly the four subjects, i.e., experimental evidences in infrared and Raman spectra of molecular crystal- acetanilide; the infrared and Raman spectra of collagen and bivine serum albumin, myoglobin and E.Coli.cell; the determination of lifetime of the solution in acetanilide and protein molecules, which are obtained by pump-probe spectroscopy and free-electron laser experiment, etc, respectively, and the evidences coming from specific heat in acetanilide and proteins. In these investigations we give not only experimental data but also their comparisons with theoretical results. The results obtained show that there are always the two peaks at 1666cm-1, or 1667cm-1 and 1650 cm-1 in infrared absorption and Raman spectra of the acetanilide, collagen, bivine serum albumin and myoglobin, which correspond to the intrinsic frequencies of the exciton and solution, respectively, the changes of strength of infrared absorption with the temperature of the systems and the specific heat are coincident between the experimental and theoretical results, the peaks of infrared absorption obtained from the experimental measurements are consistent with those in theoretical calculations. These results indicate clearly that the solution excitation exists truly in these systems.
As far as the measurement of lifetime of the solution is concerned, Fenn et al., and Hamm et al., experimental results are 15±5 ps and 35ps in the acetanilide, respectively, which were obtained by the free-electron laser experiment and pump-probe techniques, respectively. However, Austin et al., got from myglobin that the lifetime of the self-trapping state is not longer. In these experiments we always observed the occurrences of nonlinear excitations and self-trapping states. Incorporating the above results in infrared absorption and Raman spectra we have enough reasons to affirm that the solution excitation is realy existent in these systems, but we cannot finally decided and guaranteed from present measurement of lifetime that the solution has a enough lifetime to finishis the bio-energy transport because we know not so far the accurate values of velocity of motion of the solution in the acetanilide and alpha-helix proteins. Meanwhlie Austin et al., got somewhat different results, which are possibly due to the peculiarity of globular conformation of myglobin, thar differs from general alpha-helix protein, we think. Therefore, we now have not any reasons to deny the real existences of the solution in the alpha-helix protein molecules. However, we affirmed from the features of motion of the solution in biological protein molecules with physiological temperature, in which the solution moves and transpots in a supersound velocity, that the experatural results of the lifetimes by Fenn et al., Hamm et al., and Austin et al., confirm the real existences of the solution and corectness of Pang’s theory of bio-energy transport in the protein molecules, instead of other.
Acknowledgements
Author would like to acknowledge the National Basic Research Program of China (“973” program) for the financial support (Grant No. 212011CB503701).
References
- Pang XF. Biophysics, The press of Univ. of Electronic Sci. Techno of China. China. 2007; 186-208.
- Schulz GE, Schirmar RH. Principles of protein molecules. Springer. 1979; 123-175.
- Davydov AS. Solitons in quasi-one-dimensionl molecular chains. Usp fiz Nauk. 1982; 138: 603-643
- Davydov AS. Quantum theory of muscular contraction. Biophys. 1974; 19: 684-691
- Davydov AS. The theory of contraction proteins under their excitation. J Theor Biol. 1973; 38: 559-569. Ref.: https://goo.gl/vT7LS3
- Davydov AS. Solitons and energy transfer along protein molecules. J Theor Biol. 1977; 66: 379-387. Ref.: https://goo.gl/o6Ujt3
- Davydov AS, Ermakov VN. Soliton generation at the boundary of molecular chain. Physica D. 1988; 32: 318-329. Ref.: https://goo.gl/eCE4Nd
- Davydov AS. Solitons in molecular systems. Phys Scrip. 1979; 20: 387-394. Ref.: https://goo.gl/QUuV3q
- Davydov AS. Biology and quantum mechanics. Pergamon. 1982; 146-169.
- Davydov AS. Solitons in molecuar systems. Reidel Publishing Comp. 1991; 24: 133.
- Davydov AS. The lifetime of molecular solitons. J Biol Phys. 1991; 18: 111-125. Ref.: https://goo.gl/k5YsVu
- Davydov AS. The lifetime of molecular solitons. J Biol Phys. 1991; 18: 111-125. Ref.: https://goo.gl/k5YsVu
- Davydov AS. Solitons, bioenergetics and the mechanism of muscle contractions. Int J Quantum Chem. 1979; 16: 5-17. Ref.: https://goo.gl/o4A7dr
- Davydov AS. Soliton motion in a one-dimensional molecular lattice with account taken of thermal oscillations. Sov Phys JETP. 1980; 51: 397-400. Ref.: https://goo.gl/Z8MyEH
- Scott AC. Dynamics of Davydov’s soliton. Phys Rev A. 1982; 26: 578-595. Ref.: https://goo.gl/zK8fZw
- Scott AC. Davydov’s soliton. Phys Rep. 1992; 217: 1-67. Ref.: https://goo.gl/ytBBjV
- Scott AC. Thevibrational structure of Davydov solitons. Phys Scr. 1982; 25: 651-658. Ref.: https://goo.gl/KMeDc2
- Scott AC. Launching a Davydov Soliton: I. Soliton Analysis. Phys Scr. 1984; 29: 279-283. Ref.: https://goo.gl/RM93bE
- Scott AC. The laser-Raman spectrum of a Davydov soliton. Phys Lett A. 1981; 86: 60-62. Ref.: https://goo.gl/nvbwbM
- Scott AC. A nonresonant discrete self-trapping equation. Phys Scr. 1990; 42: 14-18. https://goo.gl/nGbNXe
- Brown DW. Balancing the Schrodinger equation with Davydov ansatze. Phys Rev A. 1988; 37: 5010-5011. Ref.: https://goo.gl/HgGCQP
- Brown DW, West BJ, Lindenberg K. On the applicability of Hamilton’s equation in the quantum soliton problem. Phys Rev BA. 1986; 33: 4104-4109. Ref.: https://goo.gl/zNXC8z
- Brown DW, West BJ, Lindenberg K. Nonlinear density-matrix equation for the study of finite-temperature soliton dynamics. Phys Rev B. 1987; 35: 6169-6181. Ref.: https://goo.gl/qE1AHm
- Brown DW, West BJ, Lindenberg K. Davydov solitos:new results at variance with standard derivations. Phys Rev A. 1986; 33: 4110-4115. Ref.: https://goo.gl/y79sM2
- Brown DW, Ivic Z. Unifrication of polaron and soliton theories of electron transport. Phys Rev B. 1989; 40: 9876-9887. Ref.: https://goo.gl/5Ak1ac
- Bernstein LJ. Nonlinear self-trapping in a quantum dimer. Physica D. 1991; 51: 240-243. Ref.: https://goo.gl/Av13ks
- Bernstein LJ, Eilber JC, Scott AC. The quantum theory of local modes in a coupled system of nonlinear oscillators. Nonlinearity. 1990; 3: 293-323. Ref.: https://goo.gl/7omHyt
- Brizhik LS, Davydov As. Soliton excitations in one-dimensional molecular systems. Phys Stat Sol (b). 1983; 115: 615-630. Ref.: https://goo.gl/ey7Mm5
- Skrinjar MJ, Kapor DW, Stojanovic SD. Classical and quantum approach to Davydov’s soliton theory. Phys Rev A Gen Phys. 1988; 38: 6402-6408. Ref.: https://goo.gl/D7YS7u
- Sahimi M, Hiughes BD, Scriven LE, Davis HT. Critical exponent of percolation conductivity by finite size scaling. J Phys C. 1983; 16: 521-527. Ref.: https://goo.gl/nAezeV
- Yanoviskii OE, Kryachko ES. Model for orientational defects in quasi-one-dimensional ice crystals. Phys Stat Sol (b). 1988; 140: 69-81. Ref.: https://goo.gl/WUYneh
- Yomosa S. Solitary waves in one-dimensional hydrogen-bonded system. J Phys Soc Jpn. 1983; 51: 1866-1873. Ref.: https://goo.gl/eK4vkY
- Pang XF. Soliton motions in organic protein molecules, Chin. J Biochem Biophys. 1986; 18: 1-6.
- Pang XF. The analyses of solution of revised Davydov equations. J Appl Math. 1987; 10: 228-233.
- Pang XF. The features of Davydov soliton excited in protein molecules. Chin J Atom Mol Phys. 1986; 6: 275-284.
- Christiansen PL, Scott AC. Davydov's soliton revisited. Physica D. 1991; 51: 333-342. Ref.: https://goo.gl/kTYs4G
- Brizhik LS, Davydov AS. The lectrosoliton pairing in soft molecular chains. Fiz Nizk Temp. 1983; 10: 745-753.
- Davydov AS, Kislukha NI. Solitary excitations in one-dimensional molecular chains, Phys Stat Sol (b). 1973; 59: 465-470. Ref.: https://goo.gl/dd5okP
- Cruzeiro L, Halding J, Christiansen PL, Skovgard O, Scott AC. Temperature effects on the Davydov soliton. Phys Rev A. 1988; 37: 880-887. Ref.: https://goo.gl/izR6nA
- Cruzeio-Hansson L. Mechanism of thermal destabilization of the Davydov soliton. Phys Rev A. 1992; 45: 4111-4115. Ref.: https://goo.gl/CVJ4BP
- Cruzeiro-Hansson L. Finite temperature simulations of the semiclassical Davydov model. Physica D. 1993; 68: 65-67. Ref.: https://goo.gl/A7zJ9q
- Cruzeiro-Hansson L. Two reasons why the Davydov soliton may be thermally stable after all. Phys Rev Lett. 1994; 73: 2927-2930. Ref.: https://goo.gl/RsAf76
- Cruzeiro L. The Davydov/Scott model for energy storage and transport in proteins. J Bio Physics. 2009; 35: 43-55. Ref.: https://goo.gl/8PNBJc
- Cruzeiro L. Why are proteins with glutamine- and asparagine-rich regions associated with protein misfolding diseases? J Phys Condens Matter. 2005; 17: 7833-7844. Ref.: https://goo.gl/99frEX
- Cruzeiro L. Influence of the nonlinearity and dipole strength on the amide I band of protein α-helices. J Chem Phys. 2005; 123: 4909-4917. Ref.: https://goo.gl/2zgGXF
- Cruzeiro-Hansson L, Takeno S. Davydov model: the quantum, mixed quantum- classical and full classical systems. Phys Rev E. 1997; 56: 894-906. Ref.: https://goo.gl/9ZdDJK
- Cruzeiro-Hansson L. Dynamics of a mixed quantum-classical system at finite temperature. Europhys Lett. 1996; 33: 655-659. Ref.: https://goo.gl/mWHmEz
- Cruzeiro-Hansson L, Kenkre VM. Localized versus delocalized ground states of the semiclassical Holstein Hamiltonian. Phys Lett A. 1994; 190: 59-64. Ref.: https://goo.gl/DV3KK5
- Brizhik L, Cruzeiro-Hansson L, Eremko A. Influence of electromagnetic radiation on molecular solitons. J Biol Phys. 1998; 24: 19-39. Ref.: https://goo.gl/rTwg2m
- Förner W. Quantum and disorder effects in Davydov soliton theory. Phys Rev A. 1991; 44: 2694-2708. Ref.: https://goo.gl/djuogM
- Förner W. Quantum and temperature effects on Davydov soliton dynamics: Averaged Hamiltonian method. J Phys Condens Matter. 1992; 4: 1915-1923. Ref.: https://goo.gl/uHpo7n
- Förner W. Davydov soliton dynamics: temperature effect. J Phys Condens Matter. 1991; 3: 4333-4348. Ref.: https://goo.gl/wy5kfP
- Förner W. Effects of temperature and interchain coupling on Davydov solitons. Physica D. 1993; 68: 68-82. Ref.: https://goo.gl/SBJmfw
- Motschman H, Förner W, Ladik J. Influences of heat bath and disorder in the sequence of amino acid masses on Davydov soliton. J Phys Condensed Matter. 1989; 1: 5083. Ref.: https://goo.gl/vvF197
- Förner W. Multiquanta statea derived from Davydov’s D1 ansatz: I. Equations of motion for the Su-Schrieffer-Heeger Hamiltonian. J Phys Condensed Matter. 1994; 6: 9089-9151. Ref.: https://goo.gl/juHyUw
- Förner W. Davydov soliton dynamics in proteins: II. The general case. J Mol Model. 1996; 2: 103-135. Ref.: https://goo.gl/Rc5p7P
- Förner W. Davydov soliton dynamics in proteins: I. Initial states and exactly solvable special cases. J Mol Model. 1996; 2: 70-135. Ref.: https://goo.gl/xsfcc2
- Förner W. Quantum and temperature effects on Davydov soliton dynamics: II.The partial dressing state and comparisons between different methods. J Phys Condens Matter. 1993; 5: 805-821. Ref.: https://goo.gl/wGhKgL
- Förner W. Quantum and temperature effects on Davydov soliton dynamics: III. Interchain couping. J Phys Condens Matter. 1993; 5: 823-839. Ref.: https://goo.gl/Xo2ay8
- D Hofmann, J Ladik, W Forner, P Otto. Possibility of solitary waves in the base stacks of DNA. J Phys Condensed Matter. 1992; 4: 3883-3903. Ref.: https://goo.gl/ZYrsCh
- Brizhik L, Cruzeiro-Hansson L, Eremko A. Electromagnetic radiation influence on nonlinear charge and energy transfer in biosystems. J Biol Phys. 1999; 24: 223-232. Ref.: https://goo.gl/4zNuoS
- Lomdahl PS, Kerr WC. Do Davydov Solitons Exist at 300 K? Phys Rev Lett. 1985; 55: 1235- 1238. Ref.: https://goo.gl/hhLFC5
- Kerr WC, Lomdahl, PS. Quantum-mechanical derivation of the equations of motion for Davydov solitons. Phys Rev B. 1987; 35: 3629-3632. Ref.: https://goo.gl/WLpHmv
- Wang X, Brown DW, Lindenberg K. Quantum Monte Carlo simulation of Davydov model. Phys Rev Lett. 1989; 62: 1796-1799. Ref.: https://goo.gl/raSU7N
- Wang X, Brown DW, Lindenberg K. Alternative formulation of Davydov theory of energy transport in biomolecules systems. Phys Rev A. 1988; 37: 3557-3566. Ref.: https://goo.gl/bbtJ4X
- Cottingham JP, Schweitzer JW. Calculation of the lifetime of a Davydov soliton at finite temperature. Physical Review Lettes. 1989; 62: 1792-1795. https://goo.gl/iSoJkC
- Schweitzer JW. Lifetime of the Davydov soliton. Phys Rev A. 1992; 45: 8914-8922. Ref.: https://goo.gl/LJz7Yf
- Hyman JM, Mclaughlin DW, Scott AC. On Davydov’s alpha-helix solitons. Physica D. 1981; 3: 23-44. Ref.: https://goo.gl/CAALAz
- Lawrence AF, McDaniel JC, Chang DB, Pierce BM, Brirge RR. Dynamics of the Davydov model in alpha-helix protein effects of the coupling parameter and temperature. Phys Rev A. 1986; 33: 1188-2302.
- Mechtly B, Shaw PB. Evolution of a molecular exciton on a Davydov lattice at T=0. Phys Rev B. 1988; 38: 3075-3087. Ref.: https://goo.gl/RT86bi
- Macneil L, Scott AC. Lauchinga Davydov soliton. II. Numerical analysis. Phys Scr. 1984; 29: 284-287. Ref.: https://goo.gl/oL1wJ8
- Bolterauer H, Opper M. The quantum lifetime of the Davydov soliton. Z Phys B. 1991; 82: 95-103. Ref.: https://goo.gl/3JqDp6
- Eibeck JC, Lomdahl PS, Scott AC. Soliton structure in crystalline acetanide. Phys Rev B. 1984; 30: 4703-4712. Ref.: https://goo.gl/QAhLYX
- Förner W. Davydov soliton dynamics: two-quantum states and diagonal disorder. J Phys Condens Matter. 1991; 3: 3235-3252. Ref.: https://goo.gl/tT4XAh
- Takeno S. Vibron soliton in one-dimensional molecular crystal. Prog Theor Phys. 1984; 71: 395-398. Ref.: https://goo.gl/unKi8T
- Takeno S. Vibronsolitons and coherent polarization in an exactly tractable oscillator-lattice systerm. Prog Theor Phys. 1985; 73: 853-873. Ref.: https://goo.gl/1npmqk
- Takeno S. Quantum theory of vibronsoliton-coherent states of a vibron-phonon system and self-localized modes. J Phys Soc Jpn. 1990; 59: 3127-3141. Ref.: https://goo.gl/cPV5mg
- Pang XF. The properties of collective excitation in organic protein molecular system. J Phys Condens Matter. 1990; 2: 9541-9556. Ref.: https://goo.gl/JrKzgH
- Pang XF. The dynamic properties for the protein molecular systems. Acta Math Phys. 1993; 13: 437-446. Ref.: https://goo.gl/qva1XE
- Pang XF. Mossbauer effect arising from supersonic soliton motion in organic crystal. Acta Phys Sinica. 1993; 42: 1841-1852.
- Pang XF. Properties of soliton in protein molecules with nonlinear nearest neighbour interaction. Chin Sci Bulletin. 1993; 38: 1572-1583. Ref.: https://goo.gl/FXoXX8
- Pang XF. The thermodynamic properties of the solitons excited in the protein molecules. Chin Sci Bulletin. 1993; 38: 1665-1673. Ref.:
- Pang XF. Quantum-mechamical method for the soliton transported bio-energy in protein. Chin Phys Lett. 1993; 10: 437-440. Ref.: https://goo.gl/51xSQs
- Pang XF. The specific heat cause by solitons in the protein molecular. Chin Phys Lett. 1993; 10: 381-384. Ref.: https://goo.gl/L13S55
- Pang XF. Stability of the soliton excited in protein in the biological temperature range. Chin Phys Lett. 1993; 10: 573-580. Ref.: https://goo.gl/JtQYbw
- Pang XF. Influences of temperature on features of soliton excited in the biomacromolecules. Chin J Biophys. 1993; 9: 631-641.
- Pang XF. Quantum features of the soliton excited in protein molecules. Chin J Biophys. 1994; 10: 133-142.
- Pang XF. Nonlinear quantum mechanical theory. 1st, Chinese Chongqing Press. 1994; 233-279.
- Pang XF. Comment “the thermodynamic properties of α-helix protein: A soliton approach”. Phys Rev E. 1994; 49: 4747-4752.
- Pang XF. A molecular dynamic theory of ultraweak bio-photon emission in the living systems and its properties. Chin J Atom Mol. 1995; 12: 411-421.
- Pang XF. A statistical theory for the bio-photon emission of the living systems. Chin J Atom Mol. 1997; 16: 288-296.
- Pang XF. The properties of Raman Scattering resulting from solitons excited in the organic protein molecule. Acta Physical Slovaca. 1998; 48: 99-107. Ref.: https://goo.gl/UuyiMy
- Pang XF. Influence of the soliton in anharmonic molecular crystals with temperature on Mossbauer effect. Euro Phys J B. 1999; 10: 415-425. Ref.: https://goo.gl/qPMGoy
- Pang XF, Chen XR. Nonlinear vibrational energy-spectra of molecular crystals. Chin Phys. 2000; 9: 106-110.
- Pang XF. An improvement of the Davydov theory of bio-energy transport in the protein molecular systems. Phys Rev E. 2000; 62: 6989-6998.
- Pang XF. The lifetime of the soliton in the improved Davydov model at the biological temperature 300K for protein molecules. Euro Phys J B. 2001; 19: 297-308. Ref.: https://goo.gl/Qk9dcV
- Pang XF. The effect of Raman scattering accompanied by the soliton excitation occurred in the molecular crystals. Physica D. 2001; 154: 138-149. Ref.: https://goo.gl/Dg2gmp
- Pang X, Chen XR. Distribution of vibrational energy-levels of protein molecular chains. Commun Theor Phys. 2001; 35: 323-326. Ref.: https://goo.gl/su2Lbo
- Pang XF. The features of infrared absorption of protein molecules in living systems. Commun Theor Phys. 2001; 35: 763-768.
- Pang XF, Chen XR. Calculation of vibrational energy-spectra of α-Helical protein molecules and its properties. Commun Theor Phys. 2002; 37: 715-722. Ref.: https://goo.gl/TPdLzJ
- Pang XF, Luo YH. Stabilization of the soliton transported bio-energy in protein molecules in the Improved Model. Commun Theor Phys. 2004; 41: 470-476. Ref.: https://goo.gl/W5WYBd
- Pang XF, Zhang AY. Mechanism and Properties of Non-thermally Biological Effect of the Millimeter Waves. Int J Infrared Millimeter Waves. 2004; 25: 533-552. Ref.: https://goo.gl/33Gbfy
- Pang XF, Yu JF, Luo YH. Influences of quantum and disorder effects on solitons exited in protein molecules in improved model. Commun Theor Phys. 2005; 43: 367-376. Ref.: https://goo.gl/JvK2vu
- Pang XF, Zhang HW, Yu JF, Feng YP. States and properties of the soliton transported bio-energy in nonuniform protein molecules at physiological temperature. Phys Lett A. 2005; 335: 408-415. Ref.: https://goo.gl/Ja3wSy
- Pang XF. Thermal stability of the new soliton transported bio-energy under influence of fluctuations of characteristic parameters at biological temperature in the protein molecules. Int J Modern Phys B. 2005; 19: 4677-4699. Ref.: https://goo.gl/hM4oq9
- Pang XF, Feng YP. Quantum mechanics in nonlinear systems. Singapore. 1st. ed. World Scientific Publishing Co. Singapore. 2005; 471-551.
- Pang XF, Yu JF, Luo YH. Influences of quantum and disorder effects on solitons exited in protein molecules in improved model. Commun Theor Physics. 2005; 43: 367-376. Ref.: https://goo.gl/bEhx7x
- Pang XF, Zhang HW, Yu JF, Luo YH. Thermal stability of the new soliton transported bio-energy under influence of fluctuations of characteristic parameters at biological temperature in the protein molecules. Int J Modern Physics B. 2005; 19: 4677-4699. Ref.: https://goo.gl/g85P6q
- Pang XF, Zhang HW, Yu JF, Luo YH. Influences of variations of characteristic parameters arising from the structure nonuniformity of the protein molecules on states of the soliton transported bio-energy in the improved model. Int J Mod Phys B. 2006; 20: 3027-3035.
- Pang XF, Zhang HW, Luo YH. Influences of heat bath and structure disorder in protein molecules on the soliton transported bio-energy in the improved model. J Phys Condens Matter. 2006; 18: 613-627. Ref.: https://goo.gl/3S1Cyx
- Pang XF, Zhang HW, Lui MJ, Yu.JF. Influences of heat bath and structure disorder in protein molecules on the soliton transported bio-energy in the improved model,J Phys condensed matter. 2006; 18: 613-627.
- Pang XF, Zhang HW, Yu JF, Luo YH. Influences of variations of characteristic parameters arising from the structure nonuniformity of the protein molecules on swtates of the soliton transported bio-energy in the improved model. Int J Mod Phys B. 2006; 20: 3027-3036.
- Pang XF, Yu JY, Lao YH. Combination effects of structure nonuniformity of proteins on the soliton transported bio-energy. Inter J Mod Phys B. 2007; 21: 13-42. Ref.: https://goo.gl/M8CVed
- Pang XF, Liu MU. Properties of soliton-transported bgio-energy in alpha-helix protein molecules with three channels. Commun Theory Physics. 2007; 48: 369-376. Ref.: https://goo.gl/fsSQVK
- Pang XF. Influence of structure disorders and temperatures of systems on the bio-energy transport in protein molecules. Frontier of Phys in China. 2008; 3: 457-488. Ref.: https://goo.gl/JWacbL
- Pang XF, Liu MJ. Features of motion of soliton transported bio-energy in aperiodic α-helix protein molecules with three channels. Commun Theor Phys. 2009; 51: 170-180. Ref.: https://goo.gl/QdBc1L
- Pang XF. The effects of damping and temperature of medium on the soliton excited in α -Helix protein molecules with three channels. Mod Phys Lett B. 2009; 23: 71-88. Ref.: https://goo.gl/uK9kJz
- Pang XF, Lui MJ. The Influences of temperature and chain-chain interaction on features of solitons excited in α -helix protein molecules with three channels. Int J Mod Phys B. 2009; 23: 2303-2322. Ref.: https://goo.gl/myiFj1
- Pang XF, Yu JF, Liu MJ. Changes of properties of the soliton with temperature under influences of structure disorder in the α-helix protein molecules with three channels. Mol Phys 2010; 108: 1297-1315. Ref.: https://goo.gl/6yfkMt
- Pang XF. The theory of bio-energy transport in the protein molecules and its properties. Phys Life Rev. 2011; 8: 264-286. Ref.: https://goo.gl/E9Cz5i
- Pang XF. Correctness and completeness of the theory of bio-energy transport. Phys Life Rev. 2011; 8: 302-306. Ref.: https://goo.gl/sqc9Z9
- Pang XF. The investigation of properties and theory of bio-energy transport in protein molecules. Appl Phys. 2011; 1: 47-59
- Pang XF. The properties of bio-energy transport and Influence of structure nonuniformity and temperature of systems on energy transport along polypeptide chains. Prog Biophys Mol Biol. 2012; 108: 1-46. Ref.: https://goo.gl/8EhJAc
- Pang XF. The features of nonlinear excitation and energy transport in the protein Molecules. Res Rev in BioSci. 2012; 6: 160-186
- Pang XF. The mechanism and properties of bio-photon emission and absorption in protein molecules in living systems. J Appl Phys. 2012; 111: 935191-935204. Ref.: https://goo.gl/hrYyXe
- Fohlich H. Interaction of electrons withlattice vibrations. Proc R Soc London Ser A. 1952; 215: 291-298. Ref.: https://goo.gl/CgsQMy
- Spatschek KH, Mertens FG. Nonlinear coherent structures in physics and Biology. Plenum Press, New York, USA. 1994; 56-126.
- Popp FA, Li KH, Gu Q. Recent advances in biophoton research and its application. World Scientific Publishing Co. Singapore. 1993; 141-178.
- Ho MW, Popp FA, Warnke U. Bioelectrodynamics and Biocommunication. Would Scientific Publishing Co. Singapore. 1994; 87-148.
- Pang XF. Soliton physics. Sichuan Sci Techn Press. 2003. 2-180.
- Guo BL, Pang XF. Solitons. Chin Sci Press Beijing China. 1987; 4-140.
- Bullough PK, Caudrey PJ. Soliton. Springer, New York, USA. 1982; 80-160.
- Young E, Shaw PB, Whitfield GA. Asymptotic spectrum of momentum eigestates of one-dimensional polarons. Phys Rev B. 1979; 19: 1225-1229. Ref.: https://goo.gl/oNjVxP
- Venzl G, Fischer SF. Excitonic and solitonic states in one-dimensional exciton-phonon systems. J Phys Chem. 1984; 81: 6090-6095. Ref.: https://goo.gl/Xf9GHE
- Nagle JF, Mille M, Morowitz HJ. Theory of hydrogen-bonded chains in bioenergetics. Chem J Phys. 1980; 72: 3959-3971. Ref.: https://goo.gl/sRgktu
- Wanger M, Kongeter A. A Fulton-Gouterman approach to exciton localization and excitonic solitons. Chem J Phys. 1989; 91: 3036-3044. Ref.: https://goo.gl/FY7JEz
- Eremko AA. Photodissociation of Davydov solitons. Dokl Akad Nauk Ukr SSR A. 3: 52-57. Ref.: https://goo.gl/TDXJNB
- Careri GA, Gransanti A, Ruple JA. Critical exponents of photonic percolation in hydrated lysozyme, powders. Phys Rev A. 1988; 37: 2703-2705. Ref.: https://goo.gl/7abF7E
- Careri G, Gratton E, Shyamsunder E. Fine structure of the amide-I band in acetanilide. Phys Rev A. 1988; 37: 4048-4051. Ref.: https://goo.gl/wEzUqt
- Careri G, Buontempo U, Galluzzi F, Scott AC, Gratton E, et al. Spectroscopic evidence for Davydov-like solitons in acetanilide. Phys Rev B. 1984; 30: 4689-4702. Ref.: https://goo.gl/bJs3mJ
- Careri G, Buontempo U, Caeta F, Gratton E, Scott AC. Infrared absorption in acetanilide by solitons. Phys Rev Lett. 1983; 51: 304-307. Ref.: https://goo.gl/ZgLeRJ
- Careri G, Giansanti A. Deuerium effect in the dielectric losses of wheat seeds. Lett Nuovo Cimento. 1984; 40: 193-196. Ref.: https://goo.gl/xoSerq
- Eilbeck JC, Lomdahl PS, Scott AC. Soliton structure in crystalline acetanilide. Phys Rev B. 1984; 30: 4703-4712. Ref.: https://goo.gl/ZppMLi
- Scott AC, Gratton E, Shyamsunder E, Careri G. I Rovertone spectrum of the vibrational soliton in crystalline acetanilide. Phys Rev B. 1985; 32: 5551-5553. Ref.: https://goo.gl/MkAd6E
- Scott AC, Bigio IJ, Johnston CT. Polarons in acetanilide. Phys Rev B. 1989; 39: 517-521. Ref.: https://goo.gl/9MoxFi
- Careri G, Eilbeck JC. Stability of stationary solutions of the discrete self-trapping equation. Phys lett A. 1985; 109: 201-204. Ref.: https://goo.gl/7X5Q9p
- Pang XF, Chen XR. Properties of vibration energy spectra of the molecular crystal- acetanilide. Phys Stat Sol (B). 2002; 229: 1397-1404. Ref.: https://goo.gl/Y7PuYr
- Pang XF, Chen XR. The properties of nonlinear energy-spectra of acetanilide. Int J Model Phys. 2006; 20: 2505-2510.
- Pang XF, Chen XR. Vibrational energy-spectra and infrared absorption of α-helical protein molecules. Chin Phys Lett. 2002; 19: 1096-1099. Ref.: https://goo.gl/hME9to
- Pang XF, Zhang HW. The properties of energy-spectra of molecular crystals investigated by nonlinear theory. Model Phys Lett B. 2006; 20: 1923-1932. Ref.: https://goo.gl/x4h2p2
- Pang XF, Chen XR. Quantum vibrational energy-spectra of organic molecular crystalline chains crystalline acetanilide. J Phys Chem Solids. 2001; 62: 793-796. Ref.: https://goo.gl/V1PHPW
- Alexander DM, Krumbansl JA. Localized excitations in hydrogen-bonded molecular crystals. Phys Rev B. 1986; 33: 7172-7185. Ref.: https://goo.gl/AZkjEN
- Alexander DM. Analog of small Holstein polaron in hydrogen-bonded amide systems. Phys Rev Lett. 1985; 60: 138-141. Ref.: https://goo.gl/RrjreX
- SAuvajol JL, Almarirac R, Moret J, Barthes M, Ribet JL. Temperature dependence of the Raman spectrum of fully deureratede acetanilide. J Raman Spectrosc. 1989; 20: 517-521. Ref.: https://goo.gl/6Xy8F7
- Pang XF. The features of infrared absorption arising from the solutions excited in the organic protein molecules. Chin J Inf Mill Wav. 1993; 12: 377-382.
- Pang XF. The Mossbauer effects arising from the solution excitation in organic protein molecules at biological temperature. Chin J Infra Mill Wave. 16: 288-299.
- Pang XF, Nie ZL. The effects of infrared absorption of protein molecules. Chin J Atom Mol. 1997; 14: 232-241.
- Hamm P. Femtosecond IR pump-probe spectroscopy of energy localization in protein models andmodel proteins. J Biol Phys. 2009; 35: 17-30.
- Edler J, Hamm P. Self-trapping of the amide I band in a peptide model crystal. J Chem Phys. 2002; 117: 2415-2424. Ref.: https://goo.gl/nL4S9o
- Edler J, Hamm P. Two-dimensional vibrational spectroscopy of the amide I band of crystalline acetanilide: Fermi resonance, conformational substates, or vibrational self-trapping? J Chem Phys. 2003; 119: 2709-2715. Ref.: https://goo.gl/2KRyCo
- Edler J, Hamm P, Scott AC. Femtosecond study of self-trapped vibrational excitons in crystalline acetanilide. Phys. Rev. Lett. 2002; 88: 067403.1-067403.4. Ref.: https://goo.gl/Yn57z1
- Edler J, Hamm P. Spectral response of crystalline acetanilide and N-methylacetamide: vibrational self-trapping in hydrogen-bonded crystals. Phys Rev B. 2004; 69: 214301-214307. Ref.: https://goo.gl/Vf8n28
- Edler J, Pfister R, Pouthier V, Falvo C, Hamm P. Direct observation of self-trapped vibrational states in α-helices. Phy Rev Lett. 2004; 93: 106405. Ref.: https://goo.gl/LUiZG6
- Barthes M. Optical anomalies in acetanilide-Davydov solutions, localized modes, or Fermi resonance? J Mol Liq. 1989; 41: 143-150. Ref.: https://goo.gl/zf3NSH
- Woutersen S, Mu Y, Stock G, Hamm P. Hydrogen-bond lifetime measured by time- resolved 2D-IR spectroscopy: N-methylacetamide in methanol. Chem Phys. 2001; 266: 137-147. Ref.: https://goo.gl/AubvJ2
- Blanchet GB, Fincher CR. Defects in a nonlinear pseudo one-dimensional solid. Phys. Rev. Lett. 1985; 54: 1310-1313. Ref.: https://goo.gl/xvLfG8
- Johnston CT, Swanson BI. Temperature dependence of the vibrational spectrum of acetanilide: Davydov solution or Fermi couping. Chem Phys Lett. 1985; 114: 547-552. Ref.: https://goo.gl/6yV35t
- Johnston CT, Agnew SF, Eckert J, Jones LH, Swanson BI, et al. Low-frequency single-crystal raman, far-infrared, and inelastic neutron- scattering studies of acetanilide at low-temperature. J Chem Phys. 1991; 95: 5281-5286. Ref.: https://goo.gl/azQXGy
- Pang XF, Xiao HL, Cue GP, Zhang HW, Dong B. Experiment studies of properties of infrared absorption of biological tissues. Int. J Infr Mill Wave. 2010; 31: 521-532.
- Pang XF, Zhang HW. 2006. Theoretical investigation of properties of infrared absorption of α- helix protein molecules, Int. J.Infr. Mill. Wave 27:735-744.
- Xiao HL, Cai GP, Sun SQ, Pang XF. The Properties of two-dimensional infrared spectrum of collage. Chin Atom Mol Phys. 2003; 20: 211-217.
- Cai GP, Chen LL, Yang QN. The properties of spectrum of collagen and fiber feature of silicosis. Chin J Sickness of Labour-health. 1992; 10:129-132.
- Xie A, van der Meer L, Hoff W, Austin RH. Long-lived amide I vibratrional modes in Myoglobin. Phys Rev Lett. 2000; 84: 5435-5438. Ref.: https://goo.gl/iEHXCb
- Xie A, van der Meer A FG, Austin RH. Excited-state lifetimes of far-infrared collective modes in proteins. Phys Rev Lett. 2002; 28: 147-154. Ref.: https://goo.gl/9v8HU4
- Austin RH, Xie A, van derMeer L, Shinn M, Neil G. Self-trapped states in proteins. Nucl Instrum Methods Phys Res. 2003; 507: 561-563. Ref.: https://goo.gl/sUUEDZ
- Fang C, Senes A, Cristian L, DeGrado WF, Hochstrasser RM. 2006. Amide vibrations are delocalized across the hydrophobic interface of a transmembrane helix dimer. Proc Natl Acad Sci USA. 2006; 103: 16740-16745. Ref.: https://goo.gl/L7nNK1
- Hamm P, Tsironis GP. Semiclassical and quantum polarons in crystalline acetanilide. Eur Phys J Special Topics. 2007; 147: 303-331. Ref.: https://goo.gl/JW8y8y
- Austin RH, Xie A, van der Meer L, Shinn M, Neil G. Self-trapping states in proteins? J Phys Condens matter. 2003; 15: 1693-1698.
- Austin RH, Xie A, Fu D, Warren WW, Redlich B, et al. Tilting after dutch windmills: probably no long-lived Davydov solutions in proteins. J Biol Phys. 2009; 35: 91-101. Ref.: https://goo.gl/iY8woM
- Webb SJ. Laser-Raman spectroscopy of living cells. Phys Rep. 1980; 60: 201-224. Ref.: https://goo.gl/G7JqhD
- Webb SJ, Dobbs DD. Inhibition of bacterial cell growth by 136gc microwaves. Nature. 1968; 218: 374-375. Ref.: https://goo.gl/qos9qW
- Pang XF. Physical foundations of formation of bio-self-organization and the bio-photon emission in the living systems. Int J infrared and millimeter waves. 2002; 23: 365-374. Ref.: https://goo.gl/Y8yrYQ
- Pang XF. Thermally biological effects and its medical functions of the infrared rays absorbed by living systems. Int J infrared and millimeter waves. 2002; 23: 375-391. Ref.: https://goo.gl/RGWM5V
- Pang XF. Theory of bio-energy transport in protein molecules and its experimental evidences as well as applications (I). Frontiers of Physics in China. 2007; 2: 469-493. Ref.: https://goo.gl/eX77j8
- McClare CWF. Resonance in bioenergetics. Ann N Y Acad Sci. 1974; 227: 740-97. Ref.: https://goo.gl/4sVs1j
- Fann W, Rothberg L, Roberso M, Benson S, Madey J, et al. Dynamical test of Davydov-type solutions in acetanilideusing a picosecondfree-electron laser, Phys. Rev. Lett. 1990; 64: 607-610.
- Doty P, Bradbury JH, Holtzer AM. 1956. Polypeptides. iv. The molecular weight, configuration and association of poly-γ-benzyl-l-glutamate in various solvents. J Am Chem Soc. 1956; 78: 947-954. Ref.: https://goo.gl/PsVLqr
- Knox RS, Maiti S, Wu P. Search for remote transfer of vibrational energy in proteins, in Davydov’s Solution Revisited, eds. Christiansen PL, Scott, AC. Plenum, New York. USA. 1990; 401-412.
- Backus EHG, Nguyen PH, Botan V, Pfister R, Moretto A, et al. Energy transport in peptide helices: a comparison between high- and low- energy excitation. J Phys Chem B. 2008; 112: 9091-9099. Ref.: https://goo.gl/7SSZTU
- Backus EHG, Nguyen PH, Botan V, Pfister R, Moretto A, et al. Structural flexibility of a helical peptide regulates heat transport properties. J Phys Chem B. 2008; 112: 15487-15492. Ref.: https://goo.gl/arWy5Z
- Botan V, Backus EHG, Pfister R, Moretto A, Crisma M, et al. Energy transport in peptide helices. Proc Natl Acad Sci U S A. 2007; 104: 12749-12751. Ref.: https://goo.gl/ZGQQs4
- Shen YR. IEEE. J Quantum Electron. 1986; 22: 1196-201.
- Milonni PW, Eberley JH. Lasers. Wiley, New York, USA. 1988; 198-215.
- Austin RH, Beeson K, Eisenstein L, Frauenfelder H, Gunsalus I, et al. Biochemistry. 1975; 14: 5355-5373.
- Austin RH, Xie A. Picosecond IR dynamics: lessons learned. Free Electron Lasers. 1998; 407: 504-508. Ref.: https://goo.gl/NCFxAA
- Pang XF. Nonlinear quantum mechanics.1st ed., LAP Lambert Academic Publishing, Deutschland, Germany. 2012; 272-359.
- Mrevlishvil GM. Sov. Phys-Usp. 1979; 22: 433-439.
- Mrevlishvil GM, Metreveli NQ, Razmadze GZ. Thermochim. Acta. 1998; 308: 41-46. Ref.: https://goo.gl/qU8pu9
- Katok AB, Stepin AM. Approximation of Ergodic dynamic systems by periodic transformations. Dokl Akad Nauk (SSSR). 1966; 272: 978-982. Ref.: https://goo.gl/TFNhKi
- Pang XF. Proton transfer in hydrogen bonded systems and its applications. 1st ed., LAP Lambert Academic Publishing, Deutschland, Germany. 2003; 116-154. Ref.: https://goo.gl/VmDKc8