Research Article
In silico analysis and characterization of fresh water fish ATPases and homology modelling

Rumpi Ghosh, AD Upadhayay and AK Roy*
Bioinformatics Centre, College of Fisheries, CAU(I), Lembucherra, Tripura, India
*Address for Correspondence: Dr. AK Roy, Bioinformatics Centre, College of Fisheries, CAU(I), Lembucherra, Tripura, India, Email: [email protected]
Dates: Submitted: 21 September 2017; Approved: 10 October 2017; Published: 11 October 2017
How to cite this article: Ghosh R, Upadhayay AD, Roy AK. In silico analysis and characterization of fresh water fish ATPases and homology modelling. Ann Proteom Bioinform. 2017; 1: 018-024. DOI: 10.29328/journal.hpbr.1001003
Copyright License: © 2017 Ghosh R, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: ATPase; Expasy’s prot; Physicochemical characterisation; Clustal W; Modelling etc

Abstract
ATPases is known to be a crucial in many biological activities of organisms. In this study, physicochemical properties and modeling of ATPases protein of fish was analysed using In silico approach. ATPases a protein selected from fish species, including Gold fish (Carassius auratus auratus), Zebra fish (Hypancistrus zebra), White fishes (Coregonus autumnalis), Grass carp (Ctenopharyngodon idella) and Anabas testudineus (Koi) were used in this study. Physicochemical characteristics showed with molecular weight (25045.58-25148.57Da), theoretical isoelectric point (9.30-9.97), extinction coefficient(26470-34950), aliphatic index(147.31-150.35), instability index(32.84-42.67), total number of negatively charged residues and positively charged residues (5/7-6/8), and grand average of hydropathicity (1.014-1.151) were computed. All proteins were classified as transmembrane proteins. In secondary structure prediction, all proteins were composed of random coils as predominant, followed by extended strands, alpha helix and beta turn. Three dimensional structure of protein were predicted and verified as good structures. All model structures were evaluated being accepted and reliable based on structural evaluation and stereo chemical analysis.
Introduction
ATPases an enzyme that hydrolyzes ATP; especially: one that hydrolyzes ATP to ADP and inorganic phosphate called also adenosine triphosphatase. Medical Definition of ATPase an enzyme that hydrolyzes ATP; Transmembrane ATPases are membrane-bound enzyme complexes/ion transporters that use ATP hydrolysis to drive the transport of protons across a membrane. Some transmembrane ATPases also work in reverse, harnessing the energy from a proton gradient, using the flux of ions across the membrane via the ATPase proton channel to drive the synthesis of ATP. There are several different types of transmembrane ATPases, which can differ in function (ATP hydrolysis and/or synthesis), structure (e.g., F-, V- and A-ATPases, which contain rotary motors) and in the type of ions they transport. The different types include: F-ATPases (ATP syntheses, F1F0-ATPases), which are found in mitochondria, chloroplasts and bacterial plasma membranes where they are the prime producers of ATP, using the proton gradient generated by oxidative phosphorylation (mitochondria) or photosynthesis (chloroplasts). V-ATPases (V1V0-ATPases), which are primarily found in eukaryotes and they function as proton pumps that acidify intracellular compartments and, in some cases, transport protons across the plasma membrane. They are also found in bacteria. A-ATPases (A1A0-ATPases), which are found in Archaea and function like F-ATPases, though with respect to their structure and some inhibitor responses, A-ATPases are more closely related to the V-ATPases-ATPases (E1E2-ATPases), which are found in bacteria and in eukaryotic plasma membranes and organelles, and function to transport a variety of different ions across membranes’-ATPases, which are cell-surface enzymes that hydrolyse a range of NTPs, including extracellular ATP. F-ATPases (also known as ATP Synthase, F1F0-ATPase, or H (+)-transporting two-sector ATPase) are composed of two linked complexes: the F1 ATPase complex is the catalytic core and is composed of 5 subunits (alpha, beta, gamma, delta, epsilon), while the F0 ATPase complex is the membrane-embedded proton channel that is composed of at least 3 subunits (A-C), with additional subunits in mitochondria. Both the F1 and F0 complexes are rotary motors that are coupled back-to-back. In the F1 complex, the central gamma subunit forms the rotor inside the cylinder made of the alpha (3) beta (3) subunits, while in the F0 complex, the ring-shaped C subunits forms the rotor. These ATPases can also work in reverse in bacteria, hydrolyzing ATP to create a proton gradient.
Materials and Methods
Protein sequence obtained and analysis
ATP Synthase F0 proteins of different fish species, including Gold Fish (Carassius auratus auratus-Accession No. -AEH99465.1), Hypancistrus zebra (Accession No. APF31803.1), Coregonus autumnalis (Accession No. -ABO14995.1), Ctenopharyngodon idella (Accession No. - ALS20290.1), Anabas testudineus (Accession No- AQT00813.1) were retrieved from the NCBI Protein database (http:/www.ncbi.nlm.nih.gov) under the FASTA format for analysis. The above obtained sequence was further used for complete protein analysis (structure and functional annotation) and model building using comparative modelling approach. Using expasy’s protparam server (http://expasy.org/cgi-bin/protparam) complete primary structure analysis has been performed. SOPMA was used for secondary structure prediction of protein sequence.
Sequence alignment
Multiple sequence alignment between Mitochondrion ATPase sequences from different fish species was performed using the clustalW2 server (http:/www.ebi.ac.uk/tools/msa/clustalW2/). Neighbour- Joining phylogenetic analysis of protein sequences was also generated using Clustal omega [1-3]. Clustal W2 is a server for multiple sequence alignment which is also used for phylogenetic tree analysis. Phylip and mega also available server for phylogenetic tree analysis server.
Physiochemical characterization
Physiochemical properties of the proteins such as molecular weight (Mol. wt.), amino acid composition, theoretical isoelectric point (pI), total number of positive (Arg+Lys) and negative (Asp+Glu) residues (+R/-R), extinction co-efficient (EC), instability index (AI), and grand average of hydropathicity (GRAVY) of investigated proteins was analysed by searching on the Expasy’s protparam server (http://web.expasy.org/protparam/) For the domain structures the simple Molecular Architecture Research tool (SMART) program (http:/smart.embl-heidelberg.de/) was used, for primary structure analysis as expasy’s protparam server.
Functional analysis
The server SOSUI (Hirokawa et al.) was performed to identify the types of protein. The CYS_REC (http://linux1.softberry.com) was used to predict the Presence or absence of disulphide bonds and their bonding pattern, which are crucial in defining the functional linkage and the stability of a protein. So, CYS_REC used to determine presence or absence of cystein bond.
Protein structure prediction
Secondary structure of proteins was predicted using SOPMA server (http://npsaprabi.ibcp.fr/cgibin/npsa_automat.pI?page=/NPSA/npsa_sopma.html), with the default parameters (window width: 17; similarity threshold: 8; number of states: [4,5]. Homology modeling was constructed using Swiss model server (http//swissmodel.expasy.org/) [6,7]. Swiss model is a server which is used for 3D structure prediction and also template selection, template is select based on maximum similarity or identity with sequence. Quality and accuracy with validation of the predicted models were analysed performing RAMPAGE for Ramachandran plot analysis [10,11]. The best selected models were based on the total number of residues in the most favoured regions, additional allowed region, generously allowed region and disallowed region as well as an overall G-factor have over 90% in most favoured region and cut off value (>-0.5) of overall G-factor [11,12]. Raptor X also available server for 3d structure prediction and PSIPRED and GOR IV also available free server for secondary structure analysis server. For protein structure prediction also available server are phyre 2, HHpred, modeller, CPH models, lomates,Modbase and Robette etc.
Results and Discussion
Physico-chemical characterisation
Amino acid composition in ATPase computed using Expasy’s prot param server. The physicochemical characterisations of proteins were obtained analysing Expasy’s Protparam tools (Table 1). The value of isoelectric point (pI) of proteins were ranged from 9.30-9.97(more than 7), implying the basic character of these proteins. The pI values function in protein purification by isoelectric point focusing on a polyacrylamide gel. Total number of positively (Asp+Glu) and negatively (Arg+Lys) charged residues (+R/-R) was ranged from 5 to 6 and 7 to 8, respectively. The extinction co-efficient (EC) of proteins measured at 280 nm was in a range of 31970 to 34950 M-1.cm-1 (assuming all pairs of cysteine residues from cysteins). The high value of ECs in this study implied a high concentration of cysteine along the protein sequences, functioning in quantitate the protein concentration in a volume of solution. The Instability index (II) value evaluates the stability of proteins in a test tube; it was recommended that a protein is stable when its II value is smaller than 40 and as unstable when such value is above 40. This study results showed that the II value proteins was in a range of proteins was in a range of 32.84-42.67 showing the protein of zebra fish is (II>40) and the rest is stable (II<40) [13-22]. The aliphatic index (AI) is a parameter for estimating thermal stability of a protein directly associating with the mole fraction of aliphatic side chains (Alanine, isoleucine, leucine and valine ) in the protein. In this study a high Aliphatic index values of proteins (147.31-150.35) imply high thermo stability of these proteins. Low grand average hydropathicity (GRAVY) regarded as a measure for the stability of globular protein at high temperature. The amino acid composition in ATP Synthase-F0 computed using expasy’s protparam was showed in table 1.
| Table 1: Parameters computed using Expasy’s Protparam tool. | ||||||||
| Species | No. of aa | Mol. wt. | pI | +R/-R | EC | II | AI | GRAVY |
| Zebra fish | 227 |
25148.57 |
9.30 | 6/8 | 31970 |
42.67 | 149.56 |
1.014 |
| Grass Carp | 227 | 25055.57 |
9.51 | 5/7 | 31970 |
32.84 |
147.31 |
1.151 |
| Autumnalis | 227 |
25055.57 |
9.51 |
5/7 | 31970 |
32.84 |
147.31 |
1.151 |
| Gold fish(auratus) | 227 | 25047.49 |
9.30 |
5/7 | 34950 |
33.16 |
150.35 |
1.135 |
Anabas testudineus |
227 | 25045.58 | 9.97 | 5/8 | 26470 |
36.10 | 147.44 |
1.080 |
All proteins were classified as transmembrane proteins through SOSUI program. The transmembrane regions predicted from protein sequences were shown in Table 2. These amino acid sequences of Membrane Protein have 6 transmembrane helices, except Coregonus autumnalis which have 5 transmembrane regions.
| Table: 2: Types of protein and transmembrane region identified by using SOSUI. | ||||
| Species | Types of protein | Length | Transmembrane region | N-C terminal |
| Grass Carp | PRIMARY | 23 | PSFLGIPLIAIAIALPWVLFPTP | 60-82 |
| SECONDARY | 23 | WALLLASLMVFLITINMLGLLPY | 117-139 | |
| PRIMARY | 20 | SLNMGFAVPLWLATVIIGMR | 148-167 | |
| PRIMARY | 23 | PLIPVLIIIETISLFIRPLALGV | 185-207 | |
| PRIMARY | 23 | TAGHLLIQLIATAVFVLLPMMPT | 214-236 | |
| PRIMARY | 23 | FLLTLLEVAVAMIQAYVFVLLLS | 246-268 | |
| Zebra fish | PRIMARY | 22 | PTFLGIPLIAIALTLPWILIPS | 55-76 |
| SECONDARY | 23 | WALILTSLMIFILSLNMLGLLPY | 112-134 | |
| PRIMARY | 20 | LSLNMGFAVPLWLATIIIGL | 142-161 | |
| PRIMARY | 23 | LIPVLIIIETISLFIRPLALGVR | 181-203 | |
| SECONDARY | 23 | HLLIQLISTATFILLPMMTTVAL | 212-234 | |
| PRIMARY | 23 | ILLTLLEVAVAMIQAYVFVLLLS | 241-263 | |
| Anabas testudineus | PRIMARY | 23 | PIFLGVPLIALALALPWILFPTP | 55-77 |
| SECONDARY | 23 | WALLFTSLMLFLMTLNMLGLLPY | 112-134 | |
| SECONDARY | 21 | LSLNMAFAVPLWLATVIIGMR | 142-162 | |
| PRIMARY | 23 | PLIPVLIIIETISLLIRPLALGV | 180-202 | |
| PRIMARY | 23 | IQLIATAAFVLLPLMPAVAILTA | 215-237 | |
| PRIMARY | 22 | IQLIATAAFVLLPLMPAVAILTA | 248-269 | |
| Carassius auratus | SECONDARY | 23 | ASPSYLGIPLIAIAIALPWVLYP | 57-79 |
| SECONDARY | 23 | WALLLASLMIFLITINMLGLLPY | 116-138 | |
| PRIMARY | 20 | SLNMGFAVPLWLATVIIGMR | 147-166 | |
| PRIMARY | 23 | PLIPVLIIIETISLFIRPLALGV | 184-206 | |
| PRIMARY | 23 | TAGHLLIQLIATAVFVLLPMMPT | 213-235 | |
| PRIMARY | 23 | FLLTLLEVAVAMIQAYVFVLLLS | 245-267 | |
| Coregonus autumnalis | SECONDARY | 23 | ISFMSPTYLGIPLIAVALTLPWI | 47-69 |
| PRIMARY | 23 | MLTSLMLFLITLNMLGLLPYTFT | 112-134 | |
| SECONDARY | 21 | QLSLNMGLAVPMWLATVIIGM | 138-158 | |
| PRIMARY | 23 | PLIPVLIIIETISLFIRPLALGV | 177-199 | |
| PRIMARY | 23 | IATAAFVLLPMMPTVAILTALVL | 215-237 | |
| In grass carp Total length: 275 A. A. Average of hydrophobicity: 0.844728, in zebra fish Total length: 270 A. A., Average of hydrophobicity: 0.754445, Anabas testudineus, Total length: 270 A. A., Average of hydrophobicity: 0.798519, Carassius auratus, Total length: 274 A. A., Average of hydrophobicity: 0.861314, Coregonus autumnalis, Total length: 246 A. A. Average of hydrophobicity: 0.841057. | ||||
Sequence alignment
A multiple amino acid sequence alignment of proteins was performed [1] (Figure 1A,B). The result indicated a high amino acid sequence similarity between the ATPases of five studied fish species; it was observed that between Coregonus and Hypancistrus was greater similarity. A neighbour-joining phylogenetic tree was constructed using clustal omega.
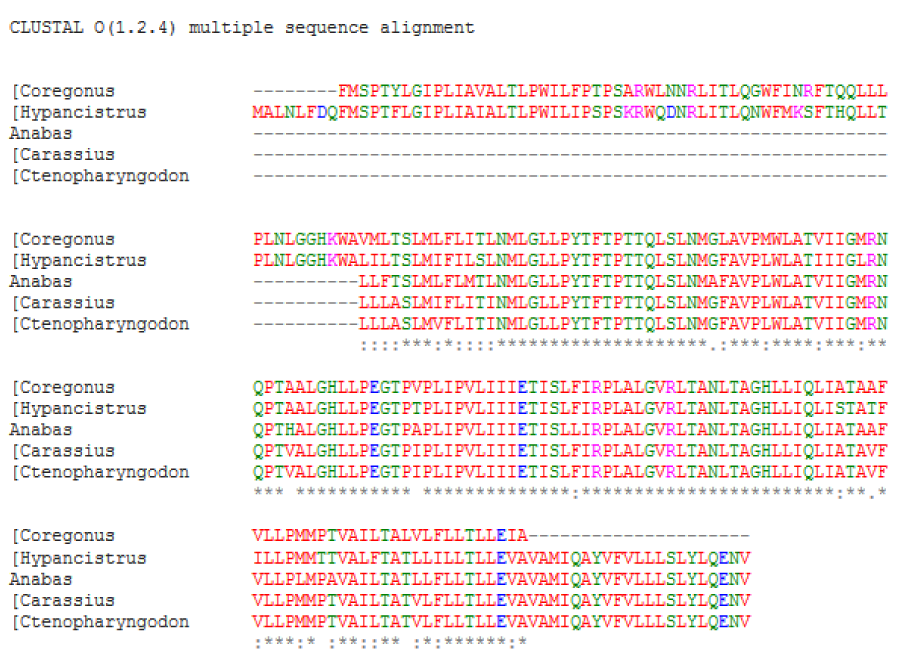
Figure 1A: Sequence alignment and phylogenetic analysis between fish ATPases sequences. (A) Multiple sequence alignment. The asterisk marks (*), colon (:), dot (.) and dash (-) indicated identical amino acids, conserved substitutions, semi conservative and deletions, respectively.
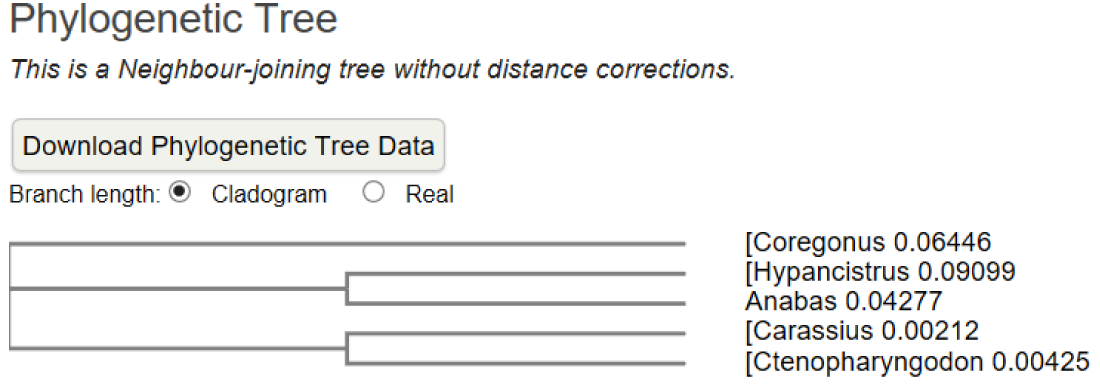
Figure 1B: A neighbour-joining phylogenetic tree showing the relationships between the ATPases.
Functional analysis result
Disulphide bonds are significant in the protein folding and stability, which are generated between the thiol groups of cysteine residues by oxidative folding process. In this study, the cysteine residues in the proteins were determined using CYS_REC server. The results revealed that any of these proteins not contain cysteine residues and most probable patterns of pairs of cysteine were not found (Table 3), suggesting that no one proteins contain disulphide bonds [4].
| Table 3: Disulphide bond patterns of pairs predicted using CYC_REC tool. | |
| Species | CYS_REC |
| grass carp | Nil |
| zebra fish | Nil |
| Anabas testudineus | Nil |
| Carassius auratus | Nil |
| Coregonus autumnalis | Nil |
There is no CYS_REC found in five Fish species. Cysteine residues and disulphide bonds which are important in determining the thermo stability of proteins. The results indicated the probable absence of disulphide bonds in these proteins.
Protein structure prediction and validation
The secondary structure of ATPases protein from fish species was predicted using SOPMA (Table 4). The results showed that except for ATPases from grass carp all contain alpha helix as a predominant component among the secondary structure elements, followed by random coil, extended strand and beta turn.
The three dimensional structures of fish ATPases protein were modelled based on the sequence and structural similarity to different available protein structure templates from the pdb (Table 4). The final structure of the models represented with the Swiss pdb viewer was shown in figure 2. Validation and predicted models performing Rampage for Ramachandran plots were represented in table 5 [4].
| Table 4: Secondary structure elements of ATPases of fish species using SOPMA. | |||||
| Element | Grass Carp | zebra fish | Anabas testudineus | Carassius auratus | Coregonus autumnalis |
| Alpha Helix 310 Helix Pi helix Beta Bridge Extended Strand Beta Turn Bend region Random coil Ambiguous states Other states |
113(49.78%) 0(0.00%) 0(0.00%) 0(0.00%) 36(15.86%) 7(3.08%) 0(0.00%) 71(31.28%) 0(0.00%) 0(0.00%) |
120(52.86%) 0(0.00%) 0(0.00%) 0(0.00%) 36(15.865) 7(3.08%) 0(0.00%) 64(28.19%) 0(0.00%) 0(0.00%) |
125(55.95%) 0(0.00%) 0(0.00%) 0(0.00%) 29(12.78%) 11(4.85%) 0(0.00%) 60(26.43%) 0(0.00%) 0(0.00%) |
111(48.90%) 0(0.00%) 0(0.00%) 0(0.00%) 39(17.18%) 11(4.85%) 0(0.00%) 66(29.07%) 0(0.00%) 0(0.00%) |
92(46.46%) 0(0.00%) 0(0.00%) 0(0.00%) 37(18.69%) 6(3.03%) 0(0.00%) 63(31.82%) 0(0.00%) 0(0.00%) |
| Table 5: Homology modeling of three –dimensional (3-D) structures of ATPase protein of fish species using SWISS MODEL and Rampage for Ramachandran plot analysis. | |||||
| Index | Grass carp | zebra fish | Anabas testudineus | Carassius auratus | Coregonus autumnalis |
| Template | 5arh.1.V | 5ara.1.V | 5ara.1.V | 5are.1.V | 5ara.1.V |
| Resolution (Å) | 7.2 | 7.2 | 6.7 | 7.4 | 6.7 |
| Sequence identity (% ) | 62.42 | 53.49 | 60.51 | 55.38 | 52.31 |
| Q mean | -3.71 | -5.68 | -3.82 | -4.37 | 5.34 |
| Rampage | |||||
| Residues in most favoured region | 4064(88.8%) | 4047(88.4%) | 4047(88.4%) | 4050 (88.5%) | 4050(88.5%) |
| Residues in generously allowed region | 351(7.7%0 | 347(7.6%) | 347(7.6%) | 351 (7.7%) |
351(7.7%) |
| Residues in out lier region | 161(3.5%) | 182(4.0%) | 182(4.0%) | 175 (3.8%) |
175(3.8%) |

Figure 2: Structure of the models represented with the Swiss pdb viewer of different fish protein.
The stereo chemical quality and accuracy of proposed models were examined performing PROCHECK analysis shown in table 4. The analysis results revealed that the predicted models for ATPase of Grass Carp, zebra fish, Anabas testudineus, Carassius auratus, and Coregonus autumnalis have over (88%) of residues in the most favoured region, indicating that these homology models were good quality and additional allowed regions combined, implying acceptable. Results showed that over 88% (88.8%, 88.4%, 88.4%, 88.5%, and 88.6% respectively) of residues found in the most favoured regions. More over 7% (7.7%, 7.6%, 7.6%, 7.7%, 7.7%) of residues in the generously or additional allowed regions, and (3.5,4.0,4.0,3.8,3.8)% residues in the disallowed regions of the proteins. All protein models contained the lower than 8% fell of residues in the generously allowed regions, indicating that may be near to be good quality models. Q-mean score value range (-5.68-5.34), the result implied that models were accepted.
Conclusion
In this study, five ATPase proteins of freshwater fish species were selected to characterise using computational tools. Physicochemical and functional characterisations of the proteins were profoundly investigated. All proteins were classified as transmembrane protein, an approximately number of alpha helix and random coils were computed to be dominating, followed by extended strands in the secondary structure of all proteins. The three dimensional models of proteins was predicted and validated by the accuracy of ramachandran plot analysis; the results suggested that all proposed models are reliable and valid. This study provide information on the physiochemical characteristics, structural properties and molecular functions of fish ATPase, which are useful for further studies on specific functions.
Acknowledgement
Authors are thankful to the dean, college of fisheries, central Agriculture University, Lembucherra, Agartala for encouragement and support. The financial assistant by DBT, GOI, New Delhi, India for BIF project under which this study has been carried out, is duly acknowledged.
References
- Tran Ngoc Tuan, Pham Minh Duc. In silico analysis of freshwater fish major histocompatibility complex Class II Alpha. Asia pacific journal of science and technology. 2016; 21: 1-8. Ref.: https://goo.gl/MtFwo3
- Tran Ngoc Tuan, Wang Gui-Tang, Pham Minh duc. Tumor necrosis factor alpha of teleosts: In silico characterization and homology modeling. Songklanakarin Journal of Science & Technology. 2016; 38: 549-557. Ref.: https://goo.gl/YB3dgD
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Molecular biology and Evolution. 2013; 30: 2725-2729. Ref.: https://goo.gl/PYMja7
- Hogg PJ. Disulfide bonds as switches for protein function. Trends Biochem Sci. 2003; 28: 210-214. Ref.: https://goo.gl/5EevyN
- Geourjon C, Deleage G. SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Computer application in bioscience. 1995; 11: 681-684. Ref.: https://goo.gl/wUsYbn
- Schwede T, Kopp J, Guex N, Peitsch MC. Swiss-MODEL:An automated protein homology modeling server. Nucleic acids research. 2003; 31: 3381-3385. Ref.: https://goo.gl/MjY825
- Arnold K, Bordoli L, Kopp J, Schwede T. The Swiss-Model workspace: A web-based environment for protein structure homology modelling. Bioinformatics. 2006; 22: 195-201. Ref.: https://goo.gl/tTxBtk
- Fiser A. Template-based Protein structure modeling. In computational Biology. 2010; 73-94.
- FiserA. Template-based protein structure modeling. D Fenyo. Computational Biology: Humana press; 2004.
- Ramachandran GN, Ramakrisnan C, Sasisekhran V. Stereochemistry of polypeptide chain configurations. J Mol Biol. 1963; 7: 95-99. Ref.: https://goo.gl/BaHh8q
- Laskowski RA, Rullman JAC, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK_NMR: Programs for checking the quality of protein structures solved by NMR. Journal of bimolecular NMR. 1996; 8: 477-486. Ref.: https://goo.gl/7Emfju
- Ramachandran G, Ramakrishnan c, Sasisekharan V. Stereochemistry of polypeptide chain configurations. J Mol Biol. 1963; 7: 95-99. Ref.: https://goo.gl/W7vzZ9
- Wallner B, Elofsson A. Can correct protein models be identified? Protein science. 2003; 12: 1073-1086. Ref.: https://goo.gl/VrWtw2
- Wiederstein M, Sippl MJ. ProSA-web: Interactive web service for the recognition of errors in three dimensional structures of proteins. Nucleic Acids Res. 2007; 35: W407-W410. Ref.: https://goo.gl/QFtmm8
- Sippl MJ. Recognition of errors in three dimensional structures of proteins. Proteins: structure, function and genetics. 1993; 17: 355-362. Ref.: https://goo.gl/cqfoDW
- Yadav NK, Shukla P, Parrek S, Singh R. Structure prediction and functional characterization of matrix protein of human Metapneumovirus (Strain can97-83)(hMPV). International Journal of advanced Life sciences. 2013; 6: 538-548. https://goo.gl/GDGdsz
- Ghosh R, Upadhayay AD, Roy AK. In silico Analysis, structure modelling and Phosphorylation site prediction of Vitellogenin Protein from Gibelion catla. Journal of applied Biotechnology and bioengineering. 2017; 3: 55.
- Pradeep N, Anupama A,Vidyashree K, Lakshmi P. In silico characterization of industrial important cellulases using computational tools. Advances science and Tecnology. 2012; 48: 14.
- Rumpi G, Upadhyay AD, Roy AK, Samik A. Structural analysis of cytochrome C Genes of Major Carp and utility of Forensic Investigation. Journal of Forensic and crime investigation. 2017; 1: 103. Ref.: https://goo.gl/ALvudA
- Tuan TN, WeiMin W, Duc PM. Characterization and homology modeling of finfish NK-kappa B inhibitor alpha using In silico analysis. J Sci Develop. 2015; 13: 216-225. Ref.: https://goo.gl/gqRHkP
- Ghosh R, Upadhayay AD, Roy AK, Mamta singh. In silico Analysis and 3d Structure Prediction of mitochondrial RHO GTPase 2 Protein of Danio rerio (zebra fish) by Homology Modeling. Jacobs J Bioinform Proteom. 2016; 1: 006. Ref.: https://goo.gl/VPmbwg
- Ghosh R, Upadhayay AD, Roy AK. In silico analysis of Rag 1 Protein of Labeo calbasu. International Journal of Innovative Research in Science & Engineering. 4: 136. Ref.: https://goo.gl/Wmo4w8