Short Communication
Function Prediction of Proteins from their Sequences with BAR 3.0

Giuseppe Profiti1,2, Pier Luigi Martelli2 and Rita Casadio2*
1ELIXIR-IIB, National Research Council, Italy
2Bologna Biocomputing Group, University of Bologna, Italy
*Address for Correspondence: Dr. Rita Casadio, Bologna Biocomputing Group, University of Bologna, Italy, Tel: +39 3495577461; Email: [email protected]
Dates: Submitted: 06 June 2017; Approved: 21 June 2017; Published: 23 June 2017
How to cite this article: Profiti G, Martelli PL, Casadio R. Function Prediction of Proteins from their Sequences with BAR 3.0. Ann Proteom Bioinform. 2017; 1: 001-005. DOI: 10.29328/journal.apb.1001001
Copyright License: © 2017 Profiti G, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

SUMMARY
Protein functional annotation requires time and effort, while sequencing technologies are fast and cheap. For this reason, the development of software tools aimed at predicting protein function from sequences can help in protein annotation.
In this paper, we describe how to use our recently implemented Bologna Annotation Resource (BAR) version 3.0, a tool based on over 30 million protein sequences for protein structural and functional annotation. In BAR 3.0, sequences are arranged in a similarity graph and then clustered together when they share at least 40% sequence identity over 90% of sequence alignment, for a total of 1,361,773 clusters.
Protein sequences with known function transfer their annotation to other sequences in the same cluster after statistical validation. Sequences with unknown function and new sequences entering in a cluster inherit its statistically validated annotations.
The method well compares to other techniques in the Critical Assessment of protein Function Annotation algorithms (CAFA). The CAFA experiment tests the performances of different predictors on a dataset that accumulates annotations over time. BAR predictions have been submitted to all the instances of CAFA through the years (BAR Plus in CAFA, BAR++ in CAFA2 and BAR 3.0 in CAFA3). The benchmarking indicates that in the field improvement is still possible and that our BAR scores among the top performing methods.
This work focuses on how the tool can transfer statistically significant features to poorly annotated or new sequences derived from transcrptomics or proteomics experiments.
INTRODUCTION
Cheap and fast sequencing technologies are widespread, and they constantly produce a large volume of biosequence data (DNA, RNA, Proteins). Protein sequences are stored in the reference UniProtKB database [1]. Then, the attribution of structural and functional features to a protein sequence (the annotation process) starts. Structural and function features are evaluated using experimental techniques that require time and different available technologies. This has been promoting a huge gap between the number of protein sequences whose biochemical and structural characteristics are documented and the vast majority of deposited sequences (presently some 85 millions). It is worth considering that more than 60 million protein sequences are labelled as "predicted” in UniprotKB (http://www.uniprot.org/statistics/TrEMBL). To overcome the gap, sequences are filtered with bioinformatics tools specifically suited to predict functional and structural features. The tools exploit the available knowledge to infer properties of the new sequences, using different approaches like machine learning and similarity searches [2,3].
The system we developed for protein functional annotation is the Bologna Annotation Resource (BAR) [4-7]. The method transfers statistically validated annotation thanks to a clustering mechanism, based on strict similarity requirements. BAR is built on a graph representation of the sequence space from UniprotKB: each protein sequence is a node, and edges represent pairwise similarity. Only edges representing a sequence identity of at least 40% over 90% of the alignment length are kept. Connected nodes are then grouped into the same cluster.
After identifying clusters, Gene ontology (GO) [8], and PFAM (PFAM) [9], annotations that are protein associated in UniprotKB, are statistically validated to identify over-represented terms. Statistical validation is performed via a Bonferroni-corrected Fisher test, and validated terms that become cluster specific are spread to all the sequences in the cluster. Protein Data Bank (PDB) [10], structures associated to proteins in a cluster, after structural alignment, are used to build structural models for sequences inside a given cluster.
The 2011 version of BAR (BAR Plus) predictions were validated by the Critical Assessment of protein Function Annotation algorithms (CAFA), reaching top scores when compared to over 50 state-of-the-art methods [2]. The 2013 version (BAR++) showed a good performance for some targets, highlighting the need for an update [3]. The present version (BAR 3.0) is both an update and an improvement on the functionalities of the system. Prediction quality was tested on the CAFA2 dataset [3]: BAR 3.0 performances has been compared to the previous version and state-of-the-art techniques [7]. The new version performs at the state art in all the Gene Ontology branches.
Furthermore, new features of the system include information about KEGG pathways [11], and cross-cluster links, with protein-protein interactions from IntAct [12], and physical interaction of protein complexes. Another improvement is the possibility to query not only by sequence, but also by annotation. We would like to propose BAR 3.0 as a useful tool for protein annotation in trascriptomics and proteomics experiments.
THE METHOD
BAR 3.0 [7], contains 32,268,689 sequences either grouped in clusters or isolated as singletons. There are 28,869,663 sequences in 1,361,773 clusters, while 3,399,026 are singletons. 97% of SwissProt sequences fall in clusters, allowing the transfer of annotation.
Statistical validation of annotation led to 674,431 clusters having some validated annotation. These clusters contain 25,447,079 sequences, about 88% of all clustered sequences in BAR 3.0.
About 39% of sequences are in clusters with statistically validated GO terms, PFAM families and a PDB structure. What is really important is that 11,206,902 of UniprotKB sequences get a statistically validated annotation they did not have previously.
Singletons, on the other hand, mostly lack any type of annotation: 43% of them are not associated even to electronically transferred annotations and may offer a subset of proteins that deserve some attention in terms of experimental approaches.
While performances of previous BAR versions have been benchmarked by CAFA and CAFA2 experiments [2,3]; BAR 3.0 predictions are still under assessment by the CAFA3 committee. We tested BAR 3.0 on the CAFA2 targets that accumulated experimental annotation between January 2014 and September 2014 and found that on this set BAR 3.0 scores similar or outperforms other state of the art methods [7]. The number of correctly predicted (true positive), wrongly assigned (false positive) and wrongly unassigned (false negative) terms are shown in table 1. A comparison with the state-of-the-art methods is listed in a recent paper [7].
| Table 1: Prediction statistics for Gene Ontology terms. | ||||
| GO branch | True Positive (TP) | False Positive (FP) | False Negative (FN) | F1 score |
| Biological Process | 7790 | 26156 | 12465 | 0.35 |
| Cellular Component | 4063 | 8381 | 3364 | 0.43 |
| Molecular Function | 2099 | 3449 | 840 | 0.54 |
When a new sequence is pasted in the query page (bar.biocomp.unibo.it), the alignment towards the BAR database allows (or not) entering a given annotated cluster. Entering is constraint by the alignment result (a match with a sequence in the cluster of at least 40% Identity over 90% of the alignment coverage). Upon insertion in the cluster, the sequence inherits all the statistically validated annotation (Figure 1).
RESULTS AND DISCUSSION
Users of BAR 3.0 can access the annotations using different approaches. The most common one would be to search for a UniprotKB saccession or entering a sequence in FASTA format. In this case, the query sequence is aligned against the ones already present in the system. The cluster or singleton that contains the matching sequence or any sequence that shares at least 40% sequence identity over 90% of sequence alignment is returned, if any. The information page contains statistics about the cluster: number of sequences, average length and taxonomic domains. Structural information is shown as a list of PDB, when present, associated to sequences in the cluster. For each PDB chain, ligand/s is/are also specified. A Hidden Markov Model (HMM) derived from the structures in the cluster can be downloaded from this section and adopted to model the protein structure. The alignment of the query sequence against the cluster HMM is available in PIR format, to be used with common modelling tools. When the PDB chain forms a complex with another one falling in a different cluster, such physical interaction is indicated, allowing navigation across different clusters.
Interaction and cross-cluster information is derived from IntAct protein-protein interactions. When a sequence in the cluster is marked as interacting with another one, both are listed in the "Protein-protein interactions” section, along with their respective clusters. The same section indicates when the organism of the query sequence is present in cluster containing the interacting sequence.
Gene Ontology annotations comprise the three main branches: Biological Process, Molecular Function and Cellular Component. For each GO Term, its p-value and distance from the ontology root are computed. PFAM domains are also associated to a p-value.
Information about pathways involving sequences in the cluster is presented in the "KEGG Pathways” section. As an example (Figure 1), we may consider a human un-reviewed sequence in UniprotKB, with "evidence at protein level”, with a submitted name of "Medium-chain-specific acyl-CoA dehydrogenase, mitochondrial” (B7Z9I1). It falls into BAR cluster #6075 that contains 32355 sequences, 68 of which from SwissProt. Sequences in this cluster are from over 4000 different species, comprising 176 Archaea, 504 Eukaryotes and 3755 Bacteria. The cluster contains 57 sequences with PDB structures, four of which form complexes with PDB associated to other clusters. There are also 6 known interactions of proteins from this cluster. For GO terms, there are 132 validated Biological Processes, 29 Molecular Functions and 32 Cellular Components. BAR 3.0 transfers a more specific Biological Process GO term with respect to the one electronically assigned by InterPro (GO:0033539, fatty acid beta-oxidation using acyl-CoA dehydrogenase), and it suggests possible new specific Molecular Function terms for dehydrogenase activity. Cellular Component experimentally assigned matches the prediction of BAR 3.0 (mitochondrion). With the cluster HMM, it is possible to model a 3D structure for the sequence. One of the known interactions is associated to Q92947, also a human dehydrogenase, suggesting possible interactions also for the query sequence.
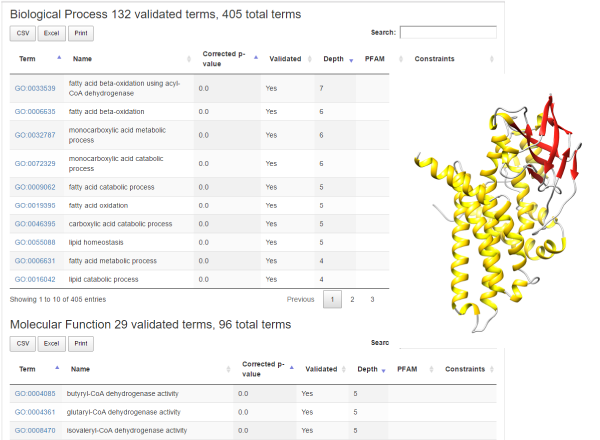
Figure 1: Inherited GO terms and 3D structure for human sequence B7Z9I1.
Besides offering a statistically validated annotation system, BAR 3.0 offers a unique opportunity for users to query for specific annotation terms (GO, PFAM, PDB), for ligands and for organisms. These searches return a list of all the clusters containing the query term. For GO terms and PFAM, clusters associated to the term in a statistically validated way are listed. For PDB, ligand and organism, all the clusters containing a sequence associated to the query term are shown. The result is presented as a table, where each row contains information about a cluster: number of sequences, number of PDB, number of validated GO terms (per branch), number of validated PFAM. If the query term was a GO or PFAM, also the associated p-value is available.
The list of resulting clusters can be narrowed further by entering a taxonomy identifier: in this way, the user can look for clusters containing a specific term and sequences from a specific organism. From the list, annotation pages for each cluster can be reached.
BAR compliments any other annotation page of the sequence if available, particularly for poorly annotated and predicted sequences, with the possibility of linking information across different clusters and fully understand the role of the sequence in the cell complex landscape.
ACKNOWLEDGEMENTS
Thanks are due to Funding for open access charge of the University of Bologna [RFO] delivered to PLM and RC. G.P. thanks ELIXIR-IIB and ELIXIR Europe for supporting his research.
REFERENCES
- UniProt Consortium. UniProt: A hub for protein information. Nucleic Acids Res. 2015; 43: 204-212. Ref.: https://goo.gl/YrmgUA
- Radivojac P, Clark WT, Oron TR, Schnoes AM, Wittkop T, et al. A large-scale evaluation of computational protein function prediction. Nat Meth. 2013; 10: 221-227. Ref.: https://goo.gl/Xg6dfK
- Jiang Y, Oron RT, Clark TW, Bankapur RA, D’Andrea D, et al. An expanded evaluation of protein function prediction methods shows an improvement in accuracy. Genome Biology. 2016; 17: 184. Ref.: https://goo.gl/LQhGpN
- Bartoli L, Montanucci L, Fronza R, Martelli PL, Fariselli P, et al. The Bologna annotation resource: a non hierarchical method for the functional and structural annotation of protein sequences relying on a comparative large-scale genome analysis. J Proteome Res. 2009; 8: 4362-4371. Ref.: https://goo.gl/DLrVmk
- Piovesan D, Martelli PL, Fariselli P, Zauli A, Rossi I, et al. BAR-PLUS: the Bologna Annotation Resource Plus for functional and structural annotation of protein sequences. Nucleic Acids Res. 2011; 39: 197-202. Ref.: https://goo.gl/9it5MU
- Piovesan D, Martelli PL, Fariselli P, Profiti G, Zauli A, et al. How to inherit statistically validated annotation within BAR+ protein clusters. BMC Bioinformatics. 2013; 3: 4. Ref.: https://goo.gl/ZM9Buz
- Profiti G, Martelli PL, Casadio R. The Bologna Annotation Resource (BAR 3.0): improving protein functional annotation. Nucl Acids Res. 2017. Ref.: https://goo.gl/gvWSiw
- Gene Ontology Consortium. Gene Ontology Consortium: going forward. Nucleic Acids Res. 2015; 43: 1049-1056. Ref.: https://goo.gl/kW74s7
- Finn RD, Coggill P, Eberhardt RY, Eddy SR, Mistry J, et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 2016; 44: 279-285. Ref.: https://goo.gl/AVdLFi
- Rose PW, Prlic A, Bi C, Bluhm WF, Christie CH, et al. The RCSB Protein Data Bank: views of structural biology for basic and applied research and education. Nucleic Acids Res. 2015; 43: 345-356. Ref.: https://goo.gl/Az5RMF
- Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017; 45: 353-361. Ref.: https://goo.gl/zQm1iq
- Orchard S, Ammari M, Aranda B, Breuza L, Briganti L, et al. The MIntAct project-IntAct as a common curation platform for 11 molecular interaction databases. Nucl Acids Res. 2014; 42: 358-363. Ref.: https://goo.gl/gWJfTW